|
|
Over the years, this research has been powered by: 

|
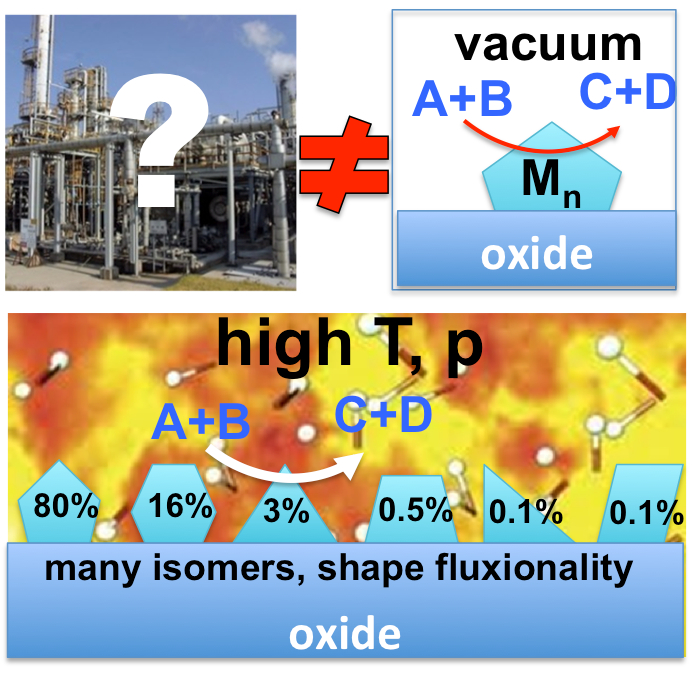
Our interest is in dynamic heterogeneous catalytic interfaces, for which we discovered a new paradigm breaking many textbook rules of catalysis, and requiring new theories and algorithms for realistic modeling. In particular, semiconductor supports decorated with sub-nano clusters of transition metals are of interest. Our group pioneered the ensemble representaiton of cluster-based catalysts: because clusters in the presence of adsorbates and at increased temperatures are dynamic, fluxional, and adopt a variety of shapes, many of these states are simultaneously populated in reaction conditiosn. Hence, the catalytic activity, selectivity, spectral characteristics, deactivation propentity, and every property indeed are ensemble-averages. As a result, many traditional rules of catalysis get broken, such as scaling relations. Importantly, ofte, it is not the most stable, but the less stable and yet thermally accessible cluster isomers are responsible for the majority of the catalytic effect. Other types of functional surfaces where this new paradigm holds up include electrocatalytic interfaces, corroding surfaces, and any kind of surface prone to the formation of defects, and reconstructions in conditions of practical use. We describe such interfaces using novel state-of-the-art algorithms that include electronic structure calculations, statistical mechanics, and elements of machine learning (such as Neural Networks). Most projects in this area are collaborative with experimentlists in surface science, catalysis, and operando spectroscopy. |

|
|
Selected References:andkarimi, B.; Poths, P.; Alexandrova, A. N. When Fluxionality Beats Size Selection: Acceleration of Ostwald Ripening of Sub-Nano Clusters. 2021, Angew. Chem. Int. Ed., 60, 11973-11982. DownloadZhang, Z.; Cui, Z.; Jimenez-Izal, E.; Sautet, P.; Alexandrova, A. N. Hydrogen Evolution on Restructured B-rich WB: Metastable Surface States and Isolated Active Sites. 2020, ACS Catal., 10, 13867-13877. Download Venegas, J.; Zhang, Z.; Agbi, T.; McDermott, W.; Alexandrova, A. N.; Hermans, I. Why Boron Nitride is such a Selective Catalyst for the Oxidative Dehydrogenaiton of Propane. 2020, Angew. Chem. Int. Ed., VIP article, 14, 16527-16535. Download Liu, G.; Poths, P.; Zhang, X.; Zu, Z.; Marshall, M.; Alexandrova, A. N.; Bowen, K. H. CO2 Hydrogenation to Formate and Formic Acid by Bimetallic Palladium-Copper Hydride Clusters. 2020, J. Am. Chem. Soc., 142, 7930-7936. Download Zhang, Z.; Zandkarimi, B.; Alexandrova, A. N. Ensembles of metastable states govern heterogeneous catalysis on dynamic interfaces. 2020, Acc. Chem. Res., 53, 447-458. Download Ha, M.-A.; Baxter, E. T.; Cass, A. C.; Anderson, S. L.; Alexandrova, A. N. Boron Switch for Selectivity of Catalytic Dehydrogenation on Size-Selected Pt clusters on Al2O3. 2017, J. Am. Chem. Soc., 139, 11568-11575. Baxter, E. T.; ha, M.-A.; Cass, A. C.; Alexandrova, A. N.; Anderson, S. L. Ethylene Dehydrogenaiton of Pt4,7,8 clusters on Al2O3: Strong Cluster-Size Dependence Linked to Preferred Catalyst Morphologies. 2017, ACS Catal, 7, 3322-3335. Zhai, H.; Alexandrova, A. N. Fluxionality of Catalytic Clusters: When It Matters and How to Address It. 2017, ACS Catal., 7, 1905-1911.
|
|
The field of quantum information science (QIS) resides in the chemical space of materials and molecules. However, this field is currently dominated by physics and engineering, and as such, fails to benefit from the geometric and electronic structure logic developed over decades in chemistry–despite the area becoming increasingly reliant on systems with increasing chemical complexity. We use the rules of chemical bonding, and tools of synthetic chemistry, spectroscopy, and theory, to bring the hitherto unrealized molecular complexity to qubit discovery, and enable substantially more flexible, scalable, and practicable quantum systems than those explored today. By leveraging chemical complexity, these QIS architectures can be tailored to meet a variety of QIS needs, from sensing to controlled chemistry. Visit our NSF Center for Chemical Innovation page: Selected references: Zhu, G.-Z.; Augenbraun, B. L.; Dickerson, C. E.; Frim, M.; Lao, G.; Lasner, Z.; Mitra, D.; Alexandrova, A. N.; Campbell, W. C.; Caram, J. R.; Doyle, J. M.; Hudson, E. R. Functionalizing Aromatic Compounds with Optical Cycling Centers. 2022, Nature Chemistry, Download Dickerson, C. E.; Guo, H.; Zhu, G.; Hudson, E. R.; Caram, J. R.; Campbell, W. C.; Alexandrova, A. N. Optical Cycling Functionalization of Arenes, 2021, J. Phys. Chem. Lett., 12, 3989-3995. Download Dickerson, C. E.; Guo, H.; Shin, A. J.; Augenbraun, B. L.; Caram, J. R.; Campbell, W. C.; Alexandrova, A. N. Franck-Condon tuning of optical cycling centers by organic functionalization. 2021, Phys. Rev. Lett., 126, 123002. Download Robinson, P. J.; Munarriz, J.; Valentine, M. E.; Granmore, A.; Drichko, N.; Chamorro, J. R.; Rosa, P. F.; McQueen, T. M.; Alexandrova, A. N. Dynamical Bonding Driving Mixed Valency in a Metal Boride. 2020, Angew. Chem. Int. Ed., VIP article, 59, 10996-110022. Download. |
|
Large protein macromolecules appear to exert meaningful intramolecular electric fields on the enzyme active sites. These fields contribute to activity and selectivity. We study these fields via quantum mechanically rigorous approaches, elucidate their specific roles in catalysis, and incorporate fields on purpose, via mutagenesis, to introduce or alter enzymatic functionality. We aim at designing catalysts that mimic natural enzymes in catalytic strategies, but catalyze reactions that interest humankind. This effort is presently fundamental. However, eventually, it might lead to unprecidented catalytic processes, green, efficient, and inexpensive, used at an industrial scale. The philosophy of this area of research is that the effort is largely done in silico, and experiment is envoked only at the end of the workflow to check theoretical predictions. Modeling of enzymes is done with atomistic and electronic insight. We develop fast techniques for mixed quantum-classical simulations and design. A particular interest is in metalloenzymes, which may containin non-physiological metals of the highest catalytic potency. At the moment we are particularly ponder the quenstion of electrostatic preorganization as one of the major driving forces behind the catalytic power of enzymes, and ways to incorportae it in our design protocols. |

|
Selected References:
Bím, D.; Alexandrova, A. N. Local Electric Fields as a Natural Switch of Heme-Iron Protein Reactivity. 2021 ACS Catal.,11, 6534-66
546. Download
|
|