
last updated Monday, March 21, 2016
A distillation is one of the most commonly used techniques to purify liquids (in some cases also solids if their boiling points are not too high
i.e., phenol, antimony(III) chloride). For instance, it is used to refine alcohol from the fermentation process or to purify water (=distilled water) or separate different hydrocarbons in raw oil (i.e., kerosine, gasolines, naphtha, etc.). In general, there are two important processes involved in this technique.
1. Vaporization = phase transfer from liquid to vapor (gas)
2. Condensation = phase transfer from vapor to liquid
The vaporization process requires heat, which makes it endothermic (ΔH>0), while the condensation process releases heat (=exothermic,
ΔH<0). The entropy increases in the vaporization process because the volume occupied from the vapor is much larger (~1000 times) compared to the liquid
(i.e., one mole of water is 18 mL of liquid but about 30600 mL at 100 °C
as steam). The opposite applies to the condensation process.
There are different forms of distillation depending on the type of compounds that are being separated from each other.
I. Simple
Distillation (in micro-scale and macro-scale)
This type of distillation is carried out at a pressure of one atmosphere (1 atm). The mixture usually only contains one volatile component i.e., salt solution, or two compounds that have boiling points that are sufficiently far away from each other i.e., diethyl ether (b.p.=34.6 oC) and toluene (b.p.=111 oC). Since many organic compounds oxidize relatively easily in air, the boiling point should be below 150 oC to avoid thermal decomposition (i.e., oxidation) of the compound during the distillation.
II. Vacuum
Distillation
This technique is usually used if the compound is either not very stable (=decomposes before its boiling point or close to it) or for compounds that have boiling points above 150 oC. For instance, N,N-dimethyl formamide (DMF), which is a commonly used solvent in organic chemistry, has a normal boiling point of 153 oC, but starts to slowly decompose into dimethylamine and carbon monoxide well below its normal boiling point. The decomposition is catatyzed by acidic or basic impurities. Therefore, the literature recommends to distill this compound below 90 oC, which requires reduced pressures (i.e., 76 oC at 30 torr).
This technique is generally not used for a low boiling solvent if a standard cooling system (=cold water) is used, because the compound does not condense anymore. An aspirator or house vacuum line is often used as source for the reduced pressure (p~30-50 torr). For a better vacuum, a rotary pump is used (p~10-2 torr). But this also requires other equipment and precautions as well. See more here.
As can be seen in the table below, the boiling points will decrease by about 60 oC for most of these compounds if the pressure is decreased from 760 torr to 40 torr. The effect is a little larger if the normal boiling point was higher like in the case of methyl benzoate. The slope of the vapor pressure as the function the temperature is different for each compound, but many compounds have similar slopes (Trouton's Rule: ΔSvap= 85-88 J/(mol*K)).
Compounds that display intermolecular hydrogen bonds (i.e., alcohols)
exhibit higher values for ΔSvap. In general, differences
in boiling point arise primarily from the different heats of vaporization.
ΔGvap=ΔHvap-Tvap*ΔSvap=0 (at the boiling point)
For compounds that exhibit a large ΔHvap, the effect is more pronounced than for compounds with low ΔHvap. Note that the enthalpy of vaporization is temperature dependent and is zero at the critical temperature. The vapor pressure of a compound can be estimated using a nomograph as shown in the SKR (page 112 (Spring 16)). A more accurate estimation can be done using the simplified Antoine equation
log(p)=A-B/(C+T)
with A, B and C being the Antoine parameters and T in ºC. The full equation contains six parameters.
|
Compound |
1
|
5 |
10 |
20 |
40 |
60 |
100 |
200 |
400 |
760 |
A |
B |
C |
ΔHvap(tb) | ΔSvap(tb) |
|
Acetic acid |
-17.2 |
+6.3 |
17.5 |
29.9 |
43.0 |
51.7 |
63.0 |
80.0 |
99.0 |
118.1 |
7.27642 |
1326.65 |
183.913 |
23.70 | 60.6 |
|
Acetone |
-59.4 |
-40.5 |
-31.2 |
-20.8 |
-9.4 |
-2.0 |
7.7 |
22.7 |
39.5 |
56.5 |
7.31414 |
1315.67 |
240.479 |
29.10 | 88.4 |
|
Acetonitrile |
-47.0 |
-26.6 |
16.3 |
-5.0 |
7.7 |
15.9 |
27.0 |
43.7 |
62.5 |
81.8 |
7.54662 |
1583.98 |
257.887 |
29.75 | |
|
Benzaldehyde |
26.2 |
51.8 |
62.0 |
76.3 |
90.1 |
99.6 |
112.5 |
132.0 |
154.1 |
179.0 |
7.25411 |
1734.80 |
217.931 |
42.50 | |
|
Benzene |
-36.7 |
-19.6 |
-11.5 |
-2.6 |
7.6 |
15.4 |
26.1 |
42.2 |
60.6 |
80.1 |
6.81404 |
1090.43 |
197.146 |
30.72 | 87.1 |
|
Benzoic acid |
96.0 |
118.6 |
132.1 |
146.7 |
162.6 |
172.8 |
186.2 |
205.8 |
227.0 |
249.2 |
7.41844 |
1824.74 |
152.886 |
50.63 | |
|
Carbon disulfide |
-73.8 |
-54.3 |
-44.7 |
-34.3 |
-22.5 |
-15.3 |
-5.1 |
10.4 |
28 |
46.5 |
7.21790 |
1303.79 |
254.394 |
26.74 | |
|
Carbon tetrachloride |
-50.0 |
-30.0 |
-19.6 |
-8.2 |
+4.3 |
12.3 |
23.0 |
38.3 |
57.8 |
76.7 |
7.01144 |
1278.54 |
232.888 |
29.82 | |
|
Chloroform |
-58.0 |
-39.1 |
-29.7 |
-19.0 |
-7.1 |
+0.5 |
10.4 |
25.9 |
42.7 |
61.3 |
7.11148 |
1232.79 |
230.213 |
29.24 | 87.5 |
|
Cyclohexane |
-45.3 |
-25.4 |
-15.9 |
-5.0 |
6.7 |
14.7 |
25.5 |
42.0 |
60.8 |
80.7 |
6.89019 |
1200.95 |
218.815 |
29.97 | 84.7 |
|
Cyclohexanol |
21.0 |
44.0 |
56.0 |
68.8 |
83.0 |
91.8 |
103.7 |
121.7 |
141.4 |
161.0 |
7.14994 |
1405.48 |
168.370 |
45.92 | |
|
1,2-Dichloroethane |
-44.5 |
-24.0 |
-13.6 |
-2.4 |
10.0 |
18.1 |
29.4 |
45.7 |
64.0 |
82.4 |
7.29525 |
1407.85 |
235.480 |
31.98 | |
|
Dichloromethane |
-70.0 |
-52.1 |
-43.3 |
-33.4 |
-22.3 |
-15.7 |
-6.3 |
8.0 |
24.1 |
40.7 |
7.11464 |
1152.41 |
232.442 |
28.06 | |
|
Diethyl ether |
-74.3 |
-56.9 |
-48.1 |
-38.5 |
-27.7 |
-21.8 |
-11.5 |
2.2 |
17.9 |
34.6 |
7.04631 |
1112.55 |
232.657 |
26.52 | |
|
Diethyl succinate |
54.6 |
83.0 |
96.6 |
111.7 |
127.8 |
138.2 |
151.1 |
171.7 |
193.8 |
216.5 |
7.88220 |
2229.64 |
228.805 |
50.57 | |
|
1,2-Dimethoxyethan |
-48.0 |
-22.0 |
-15.3 |
1.3 |
10.7 |
19.7 |
31.8 |
47.1 |
70.8 |
82 |
7.30039 |
1418.59 |
236.928 |
32.42 | |
|
Dimethylformamide |
7.2 |
31.0 |
42.6 |
55.5 |
69.9 |
79.0 |
91.5 |
110.0 |
131.0 |
153.0 |
7.24128 |
1597.92 |
213.457 |
39.41 | |
|
Dimethylsulfoxide |
32.2 |
57.8 |
70.6 |
84.7 |
100.4 |
110.3 |
123.8 |
144.0 |
166.9 |
190.9 |
7.23168 |
1733.52 |
207.580 |
43.70 | |
|
1,4-Dioxane |
-35.8 |
-12.8 |
-1.2 |
12.0 |
25.2 |
33.8 |
45.1 |
62.3 |
81.8 |
101.1 |
7.29969 |
1485.85 |
234.976 |
34.16 | |
|
Dipropyl succinate |
77.5 |
107.6 |
122.2 |
138.0 |
154.8 |
166.0 |
180.3 |
202.5 |
226.5 |
250.8 |
8.20708 |
2636.26 |
244.154 |
||
|
Ethanol |
-31.3 |
-12.0 |
-2.3 |
8.0 |
19.0 |
26.0 |
34.9 |
48.4 |
63.5 |
78.4 |
8.13484 |
1662.48 |
238.131 |
38.56 | 109.7 |
|
Ethyl acetate |
-43.4 |
-23.5 |
-13.5 |
-3.0 |
9.1 |
16.6 |
27.0 |
42.0 |
59.3 |
77.1 |
7.25963 |
1338.46 |
228.608 |
31.94 | |
|
Ethyl benzoate |
44.0 |
72.0 |
86.0 |
101.4 |
118.2 |
129.0 |
143.2 |
164.8 |
188.4 |
213.4 |
7.50855 |
2047.03 |
228.940 |
44.64 | |
|
Heptane |
-34.0 |
-12.5 |
-2.1 |
9.3 |
22.3 |
30.3 |
41.8 |
58.3 |
78.0 |
98.4 |
7.04605 |
1341.89 |
223.733 |
31.77 | 85.4 |
|
Hexane |
-53.9 |
-34.5 |
-25.0 |
-14.1 |
-2.3 |
5.4 |
15.8 |
31.6 |
49.6 |
68.7 |
6.98950 |
1216.92 |
227.451 |
28.85 | 84.4 |
|
Isopropanol |
-26.1 |
-7.0 |
+2.4 |
12.7 |
23.8 |
30.5 |
39.5 |
53.0 |
67.8 |
82.5 |
7.83056 |
1483.30 |
217.413 |
39.85 | |
|
Methanol |
-44.0 |
-25.3 |
-16.2 |
-6.0 |
5.0 |
12.1 |
21.2 |
34.8 |
49.9 |
64.7 |
8.09126 |
1582.91 |
239.096 |
25.21 | 104.3 |
|
Methyl benzoate |
39.0 |
64.4 |
77.3 |
91.8 |
107.8 |
117.4 |
130.8 |
151.4 |
174.7 |
199.5 |
7.32570 |
1825.16 |
211.583 |
44.02 | |
|
Methylethyl ketone |
-48.3 |
-28.0 |
-17.7 |
-6.5 |
6.0 |
14.0 |
25.0 |
41.6 |
60.0 |
79.6 |
7.20103 |
1325.15 |
227.093 |
31.30 | |
|
Nitromethane |
-29.0 |
-7.9 |
+2.8 |
14.1 |
27.5 |
35.5 |
46.6 |
63.5 |
82.0 |
101.2 |
7.53850 |
1588.83 |
239.919 |
33.99 | |
|
n-Pentane |
-76.6 |
-62.5 |
-50.1 |
-40.2 |
-29.2 |
-22.2 |
-12.6 |
+1.9 |
18.5 |
36.1 |
7.00877 |
1134.15 |
238.678 |
25.79 | 83.4 |
|
n-Propanol |
-15.0 |
+5.0 |
14.7 |
25.3 |
36.4 |
43.5 |
52.8 |
66.8 |
82.0 |
97.8 |
7.77374 |
1518.16 |
213.076 |
41.44 | 111.9 |
|
Pyridine |
-18.9 |
+2.5 |
13.2 |
24.8 |
38.0 |
46.8 |
57.8 |
75.0 |
95.6 |
115.4 |
7.18740 |
1463.63 |
224.598 |
35.09 | |
|
Tetrahydrofuran |
-55.8 |
-36.5 |
-26.8 |
-16.1 |
-4.3 |
3.3 |
13.6 |
29.0 |
46.5 |
64.9 |
7.10537 |
1256.68 |
232.621 |
29.81 | |
|
Toluene |
-26.7 |
-4.4 |
6.4 |
18.4 |
31.8 |
40.3 |
51.9 |
69.5 |
89.5 |
110.6 |
7.13620 |
1457.29 |
231.827 |
33.18 | 86.5 |
|
Water |
-17.3 |
+1.2 |
11.2 |
22.1 |
34.0 |
41.5 |
51.6 |
66.5 |
83.0 |
100.0 |
8.05573 |
1723.64 |
2330.76 |
40.65 | 109.1 |
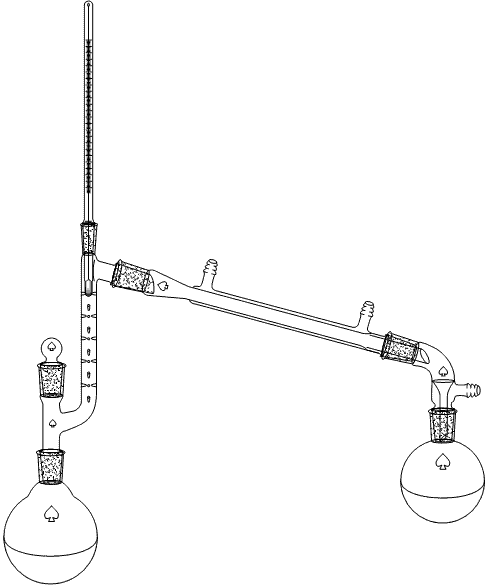
III. Fractional
Distillation
This technique is used for mixtures where the boiling points vary by less than 20 ºC from each other i.e., the big distillation setups in the petroleum industry to refine crude oil. This technique can be performed at atmospheric and reduced pressure and requires a more elaborated setup i.e., Vigreux columns (shown in diagram), spinning band columns, etc. If a mixture was distilled using a simple distillation, the boiling point of the mixture would change more gradually. If the same mixture was distilled using a fractionated distillation, the boiling point would increase dramatically after most of the low boiling component was removed. This allows for a cleaner separation of the components in a mixture. The more theoretical plates the column has, the better the separation will be.
IV. Steam
Distillation
This version is used for compounds that are insoluble in water at all temperatures, but can be co-distilled with water. This technique is often used for the isolation of natural compounds for plants i.e., eugenol from cloves, citronella from lemon grass oil, etc. It has the advantage that the maximum temperature reached this way is 100 oC, which is acceptable for most compounds, even if they do have a lot of reactive functional groups present.
The lower the boiling point of the compound, the more of the compound co-distills with the water since both, the water and the compound, contribute to the total vapor pressure of the mixture (Ptotal=PH2O + Pcompound). Unfortunately, these systems do not follow Raoult's law, meaning the composition of the vapor/distillate depends on the individual vapor pressure of the compound and not on the mole fraction of the compound in the mixture. For instance, a mixture of eugenol (b.p.= 255 ºC) and water distills at 99.5 ºC, which means that water has a vapor pressure of 747 mmHg and eugenol exhibits a pressure of 13 mmHg.
The amount of the substance X that codistills together with the water is given by
Pwater/px = nwater/nx
mwater/1 g of X = (18* Pwater)/(MMx*Px)
Using the formula above, one can deduce that an azeotropic mixture of benzene and water that boils at 69.2 ºC, is composed of 0.1 g of water and 1.0 g of benzene. The vapor pressure of water at 69.2 ºC is 227 mmHg. The amount of water codistilling with 1 g of the substance X decreases as the vapor pressure of the compound increases. For compounds
with very low vapor pressure, this procedure is not very economical
because it requires large amounts of water, thus energy.
V. Azeotropic Distillation
This techniques is often used to remove water from a reaction mixture i.e. esterification or Aldol condensation. Commonly, solvents like benzene or toluene are used to aid the removal. Water and the organic solvent co-distill and separate in a Dean-Stark trap. The water settles on the bottom of the trap, while the organic solvent runs back into the reaction mixture and removes more water. These mixtures also deviate from Raoult's law. The boiling point curves look similar to the melting poing composition curves, or just inverted, depending on the deviation from Raoult's law. An azeotrope is a constant-boiling mixture with a sharp boiling point, and a well-defined composition (similar like a eutectic mixture). The azeotrope of water and ethanol boils at 78.15 ºC and has a composition of 95.5 % of EtOH and 4.5 % of water. Other azeotropic mixtures are water:benzene (b.p.=69.2 ºC, 9:91), water:toluene (b.p.=84.2 ºC, 20:80), ethanol:benzene (b.p.=68.2 ºC, 32:68) and ethanol:benzene:water (b.p.=64.9 ºC, 19:74:7).
Since the boiling point of ethanol and the azeotrope of ethanol:water are too close, the water cannot be removed using the minimum boiling azeotrop. The addition of benzene leads to the formation of different azeotrope that possesses a boiling point significantly different from the boiling point of ethanol. First a ternary azeotrope would distill to remove the water, then the binary azeotrope to remove the benzene, leaving behind the pure ethanol in the distillation pot.
A mixture like water and formic acid form a maximum boiling
point azeotrope that boils at 107.1 ºC, while water and formic acid
boiling at 100.0 and 100.7 ºC, respectively. Hydrofluoric acid (35.6
%, 111.35 ºC), nitric acid (68 %, 120.5 ºC) and perchloric acid
(17.6 %, 230 ºC) also form a maximum boiling azeotrope.
Microscale Distillation:
The picture below shows the basic setup:
 |
The setup consists of:
1. Conical vial 2. Hickman head 3. Air condenser 4. Spin vane 5. Two big compression caps 6. Two big O-rings 7. One small compression cap (for side port of Hickman) 8. One small flat septum 9. One metal clamp 10. Wet paper towels 11. Hotplate with Al-block |
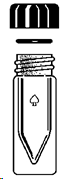
|
The conical vial should be securely attached to the Hickman head and the air condenser using the compression cap and the O-ring (shown on the right). All pieces should have a ground-glass joint (=a rough surface on the inside on the top) and fit together well (=no major leaks). The spin vane should be placed in the conical vial, point down (▼). The flat septum and the small compression cap are used to close the side port of the Hickman head. The entire setup is placed in the appropriate hole in the Al-block and centered on the hotplate before starting to stir (otherwise the spin vane will flip over and rotate improperly). The Hickman head and the air condenser have to be cooled with the wet paper towel (an intimate contact between the wet paper towel and the glassware is very important here!). The setting for the heat source should be at a level that the compound of interest distills over slowly. This way, it condenses at the walls of the air condenser and the Hickman head, and collects in the dwell of the Hickman head. If the dwell is full, the compound has to be removed using a Pasteur pipette. If the dwell is very deep, a pipette with a bent tip might have to be used in order to reach the liquid from the top.
If there is more than one volatile compound in the vial, it will be necessary to start with low heat to distill the lower boiling compound first. This way, compounds with a difference of boiling point of at least 50 ºC can be separated relatively cleanly if the distillation is performed properly.
Summary
1. The round-bottomed flask with the liquid to be distilled (=pot) should not be more than half-filled
in order to leave enough room for the liquid to boil. Otherwise the
solution will spill out or over during the boiling process.
2. A spin vane, stir bar (both have to be spinning during the heating!) or a boiling stone has to be added to avoid bumping (=spilling over).
3. A good seal between the joints minimizes the loss of target compound during the distillation. It also prevents that the compound drips onto the hotplate and catches on fire.
4. The hotplate will get extremely hot (>300 ºC) if a setting of “10” is used. It will be very difficult to control the distillation since the liquid will foam much more. An appropriate temperature setting has to be used. Often times, a water or oil bath is used for better temperature control. If a heating mantle is used as heat source, it has to be plugged into a variable power control.
5. A liquid boils when a reflux ring is going up the neck of the Hickman head or three-way adapter, and not when it starts to bubble! Often times, liquids dissolve gases that are released prior reaching the boiling point because their solubility decreases. For example, a coke can expands when you store it in a warm/hot place because the CO2 dissolved in the liquid is desorbed!
6. It is also imperative to remove the drying agent before the liquid is distilled since the drying process is reversible at higher temperatures (see chapter about Drying Solutions).
Questions:
1. Why is it imperative to leave an opening somewhere in the setup if a liquid or solid is distilled?
2. A mixture of bromobutane and water is distilled.
a. Is the boiling point of the mixture going to be above or below the lowest boiling point of the individual compounds?
b. Which of the two compounds will be the major component in the distillate in terms of moles and in terms of grams?
3. Why is toluene or benzene used in azeotropic distillations and not ethanol?
4. Compound A has a boiling point of b.p.=120 ºC, while compound B boils at b.p.=150 ºC.
a. What can be said about the vapor pressures of these compounds?
b. Would you be able to separate the two compounds using the simple distillation setup used in the lab?