last updated
Extraction (Part 1)
After a reaction is completed, the solution often times does not only contain the desired product, but also undesired byproducts of the reaction, unreacted starting material(s) and the catalyst (if it was used). These compounds have to be removed in the process of isolating the pure product. A standard method used for this task is an extraction or often also referred to as washing. Strictly speaking, the two operations are targeting different parts in the mixture: while the extraction removes the target compound from an impure matrix, the washing removes impurities from the target compound i.e., water by extraction with saturated sodium chloride solution. Washing is also used as a step in the recrystallization procedure to remove the impurity containing mother liquor adhering to the crystal surface.
Many liquid-liquid extractions are based on acid-base chemistry. The liquids involved have to be immiscible in order to form two layers upon contact. Since most of the extractions are performed using aqueous solutions (i.e., 5 % NaOH, 5 % HCl), the miscibility of the solvent with water is a crucial point as well as the compatibility of the reagent with the compounds and the solvent of the solution to be extracted. Solvents like dichloromethane (=methylene chloride in older literature), chloroform, diethyl ether, or ethyl ester will form two layers in contact with aqueous solutions if they are used in sufficient quantities. Ethanol, methanol, tetrahydrofuran (THF) and acetone are usually not suitable for extraction because they are completely miscible with most aqueous solutions. However, in some cases it is possible to accomplish a phase separation by the addition of large amounts of a salt (“salting out”). Commonly used solvents like ethyl acetate (8.1 %), diethyl ether (6.9 %), dichloromethane (1.3 %) and chloroform (0.8 %) dissolved up to 10 % in water. Water also dissolves in organic solvents: ethyl acetate (3 %), diethyl ether (1.4 %), dichloromethane (0.25 %) and chloroform (0.056 %). Oxygen containing solvents are usually more soluble in water (and vice versa) because of their ability to act as hydrogen bond donor and hydrogen bond acceptor. The higher water solubility lowers the solubility of weakly polar or non-polar compounds in these solvents i.e., wet Jacobsen ligand in ethyl acetate. Other solvents such as alcohols increase the solubility of water in organic layers significantly because they are miscible with both phases and act as a mediator. This often leads to the formation of emulsions.
The most important point to keep in mind throughout the entire extraction process is which layer contains the product. For an organic compound, it is relatively safe to assume that it will dissolve better in the organic layer than in most aqueous solutions unless it has been converted to an ionic specie, which makes it more water-soluble. If a carboxylic acid (i.e., benzoic acid) was deprotonated using a base or an amine (i.e., lidocaine) was protonated using an acid, it would become more water-soluble because the resulting specie carries a charge. Chlorinated solvents (i.e., dichloromethane, chloroform) exhibit a higher density than water, while ethers, hydrocarbons and many esters possess a lower density than water (see solvent table), thus form the top layer (see solvent table).. One rule that should always be followed when performing a work-up process:
Never dispose of any layer away until you are absolutely sure (=100 %) that you will never need it again. The only time that you can really be sure about it is if you isolated the final product in a reasonable yield, and it has been identified as the correct compound by melting point, infrared spectrum, etc. Keep in mind that it is always easier to recover the product from a different layer in a beaker than from the waste container or the sink. In this context it would be wise to label all layers properly in order to be able to identify them correctly later if necessary.
In order to separate compounds from each other, they are often chemically modified to make them more ionic i.e., convert a carboxylic acid into a carboxylate by adding a base. Standard solutions that are used for extraction are: 5 % hydrochloric acid, 5 % sodium hydroxide solution, saturated sodium bicarbonate solution (~6 %) and water. All of these solutions help to modify the (organic) compound and make it more water-soluble and therefore remove it from the organic layer. More concentrated solutions are rarely used for extraction because of the increased evolution of heat during the extraction, and potential side reactions with the solvent.
What do I use when to extract?
a. Removal of a carboxylic acid or mineral acid
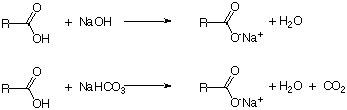
In order to remove an acidic compound from a mixture, a base like NaOH or NaHCO3 is used. The carboxylic (or mineral) acid and the base react to form a sodium salt, which is usually exhibits a higher solubility in aqueous solutions due to its negative charge and higher polarity (as indicated by a more negative log Kow value i.e., CH3COOH: -0.17, Na+CH3COO-: -3.72).
Which of the two reagents should be used depends on the other compounds present in the mixture. Sodium hydroxide is usually easier to handle because it does not evolve carbon dioxide as a byproduct. In addition, the concentration can be increased significantly if is needed. However, if compounds were present that are sensitive towards strong bases or nucleophiles (i.e., esters, ketones, aldehydes, etc.), sodium bicarbonate should be used. It does not react with these compounds because it is a weaker base and a weak nucleophile (due to its resonance stabilization). Note that the formation of carbon dioxide as a byproduct causes a pressure build-up in the separatory funnel, the centrifuge tube or the conical vial. Thus, additional precautions (i.e., frequent venting) have to be taken to prevent any accidents resulting from the pressure build up in the extraction vessel. The target compound can subsequently be recovered by adding a mineral acid to the basic extract i.e., benzoic acid in the Grignard experiment in Chem 30CL.
b. Removal of a phenol
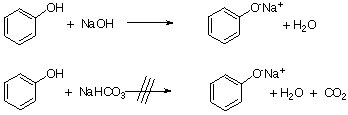
Most phenols are weak acids (pKa= ~10) and do not react with sodium bicarbonate, which is a weak base itself (pKa(H2CO3)=6.37, 10.3). However, they do react with a strong base like NaOH. This difference in acidity can be exploited to separate carboxylic acids and phenols from each other in an organic layer. While many phenols dissolve poorly in water (8.3 g/100 mL at 20 oC, log Kow=1.46), phenolates dissolve very well in aqueous solutions. After the extraction, the phenol can be recovered by adding a mineral acid to the basic extract.
c. Removal of an amine
Depending on the chain length, amines might or might not be soluble in water i.e., propylamine is miscible with water (log Kow=0.48), triethylamine displays a limited solubility at room temperature (17 g/100 mL, log Kow=1.44), while tributylamine hardly dissolves at all (0.37 g/100 mL, log Kow=4.60). Amines are basic and can be converted to ammonium salts using mineral acids i.e., hydrochloric acid. The resulting salts dissolve in water. However, the solubility of the ammonium salts decreases as the number and size of R-groups increases. Ammonium salts from primary amines are much more soluble in water than salts from tertiary amines due the increased ability to form hydrogen bonds [(H3NEt)Cl: 280 g/100 g H2O, (H2NEt2)Cl: 232 g/100 g H2O, (HNEt3)Cl: 137 g/100 g H2O (all at 25 oC)].
![]()
After separation of the organic and the aqueous layer, the amine can be recovered by addition of a strong base like NaOH or KOH to the acidic extract i.e., lidocaine synthesis. Note that amides are usually not basic enough to undergo the same protonation (pKa of conjugate acid: ~ -0.5).
d. Isolation of a neutral species
Most neutral compounds cannot be converted into salts without changing their chemical nature. Many of these neutral compounds tend to react in undesired ways i.e., esters undergo hydrolysis upon contact with strong bases or strong acids. One has to keep this in mind as well when other compounds are removed. For instance, epoxides hydrolyze to form diols catalyzed by acids and bases. Ketones and aldehydes undergo condensation reactions catalyzed by both, acids and bases. Esters also hydrolyze to form carboxylic acids (or their salts) and the corresponding alcohol. In order to separate these compounds from each other, chromatographic techniques are often used, where the compounds are separated based on their different polarities (see Chromatography chapter).
e. General Separation Scheme
Based on the discussion above the following overall separation scheme can be outlined. Which sequence is the most efficient highly depends on the target molecule. There is obviously no reason to go through the entire procedure if the compound sought after can be isolated in the first step already. Note that many of these steps are interchangeable in simple separation problems.
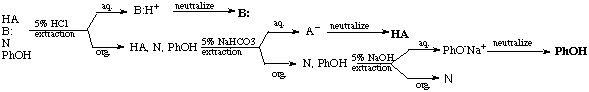
For instance, if the target compound was the base in the system, the extraction with HCl should be performed first. Whatever remains in the organic layer is not of interest anymore afterwards, unless one of the other compounds has to be isolated from this layer as well. If the target compound was an acid, the extraction with NaOH should be performed first. This strategy saves steps, resources and time, and most of all, greatly reduces waste.
Practical Aspects of an Extraction
An extraction can be carried out in macro-scale or in micro-scale. In macro-scale, usually a separatory funnel (on details how to use it see end of this chapter) is used. Micro-scale extractions can be performed in a conical vial or a centrifuge tube depending on the quantities. Below are several problems that have been frequently encountered by students in the lab:
a. Which layer is the aqueous layer?
Most solutions are relatively diluted (~5 %) and their density is not much different from that of water (i.e., 5 % HCl: 1.02 g/cm3, 5 % NaOH: 1.055 g/cm3). Thus, the density of a solid i.e., sodium hydroxide (2.1 g/cm3 in the solid) does not provide the information sought. The density is determined by the major component of a layer which is usually the solvent. About 5 % of a solute does not change the density of the solution much. However, this can change if very concentrated solutions are used (see table in the back of the reader)! Thus, diethyl ether and ethyl acetate, which are both less dense than the dilute solutions that are usually used for extraction, form the top layer, while dichloromethane and chloroform form the bottom layer (currently both of them are not used in Chem 30BL or Chem30CL due to safety concerns!).
b. Why are three layers observed sometimes?
It is not uncommon that a small amount of one layer ends up on top of the other. Mixing with a stirring rod or gentle shaking usually takes care of this problem. Small amounts (compared to the overall volume of the layer) should be discarded here.
c. Why do the layers not separate?
This would usually happen if the mixture was shaken too vigorously. Subsequently, an emulsion is formed instead of two distinct layers. In such an event, the mixture can be stirred slowly with a glass rod to bring the small droplets together a little faster, which ultimately leads to the formation of a new layer. In some cases, a careful draining of the existing lower layer can also be helpful because it pushed the bubbles together in the smaller part of the extraction vessel. In cases, where the phases have similar polarity or density, the addition of more solvent can assist the separation. Sometimes, the addition of a salt (or salt solution) can also lead to a better phase separation (“salting out”). In many cases, centrifugation or gravity filtration works as well. When it is known, through experience, that some mixtures may form emulsions, vigorous shaking should be avoided. Instead, gently rocking the separatory funnel back and forth for 2-3 minutes will accomplish sufficient degree of mixing while minimizing the formation of emulsions.
d. How do we know that we are done extracting?
Strictly speaking, hardly ever all of the solute will be extracted since there is finite distribution coefficient for the compound (see also Extraction II). As a general rule, multiple extractions with small quantities of solvent or solution are more efficient than one extraction using the same amount of solvent (see below). The amount of material left behind after two or three extractions is usually very small (less than 5 % in most cases) and does not justify the effort and resources (solvent and time to perform the extractions and to remove the solvent later on). Excessive washing will also lower the yield of the product, if the desired compound dissolves noticeably in the other phase.
e. Why does the extraction container (vial, centrifuge tube, separatory funnel) make funny noises?
This phenomenon will often be observed if sodium bicarbonate is used for the extraction in order to neutralize or remove acidic compounds. The reaction affords carbon dioxide (CO2), which is a gas at ambient temperature. Pressure builds up that pushes some of the gas and the liquid out. The container should be vented immediately before the pressure build-up can cause an explosion, an ejection of the stopper on the top or excessive spillage upon opening. A similar observation will be made if a low boiling solvent is used for extraction. The shaking of the mixture increases the surface area, and therefore the apparent vapor pressure of the solvent. In addition, many extraction processes are exothermic because they involve an acid-base reaction.
f. The centrifuge tube leaks
Often times the cap is either the wrong cap in the first place or it is not properly placed on the top. If NaHCO3 is used for extraction, the centrifuge tube has to be vented more frequently.
g. The separatory funnel leaks
Before using the separatory funnel, the user should check if the stopcock plug and the stopcock fit together well. In addition, the stopper on the top has to fit into the joint on the top to prevent leakage there (for more details at the end of this chapter).
h. Why is a centrifuge tube, a conical vial or a separatory funnel used for the extraction and not a beaker or test tube?
The conical shape of these pieces of equipment makes it easier to collect the solution on the bottom using a Pasteur pipette because of the smaller interface. The task of getting a clean phase separation will be more difficult if the liquids are spread out over a large, flat or curved surface.
i. Which layer should be removed, top or bottom layer?
The bottom layer is always removed first independently if this is the one of interest or not because it is much easier to do. If a centrifuge tube or conical vial was used, the bottom layer should be drawn using a Pasteur pipette. From this point of view, a solvent with higher density than water would be preferential, especially when very small quantities are used. This will allow to minimize the number of transfer steps required.
j. How much solvent/solution is used for the extraction?
This highly depends on the quantity of a compound that has to be removed. For most washing processes, 10-20 % of the volume of the solution to be washed will do an adequate job. If a large amount of a compound has to be transferred or neutralized, more concentrated solutions and larger quantities might be needed. Multiple extractions with smaller quantities are preferred over one extraction with the same quantity of solution/solvent. If solutions with higher concentrations are used, extra caution is advised because neutralization reactions are exothermic. This can pose a serious problem when using low boiling solvents i.e., diethyl ether, dichloromethane, etc. because a pressure build-up will be observed in the extraction container.