![]() ,
, ![]()
![]() masses of involved atoms
masses of involved atoms
F=force constant
last updated
Infrared Spectroscopy
Dr.A.Bacher
1. Theory
Vibrational modes within a molecule can be described using the anharmonic oscillator model. This model assumes that the two masses (with known weight) are connected with a spring (with known strength). With the help of quantum mechanical calculations (Schroedinger equation) you can find the frequencies of basic stretching and bending modes:
![]() ,
, ![]()
![]() masses of involved atoms
masses of involved atoms
F=force constant
From this equation, one can deduce some basic trends can be deducted:
a. If the force constant F (= bond strength) increases, the stretching frequency will increase as well (in cm-1)
| CC bond | triple | double | single |
| 2100 | 1650 | 1130 | |
| functional group | alkyne | alkene | alkane |
b. If the masses of the involved atoms increase, the peak will shift to lower wavenumbers e.g. H/D-exchange in labeling experiments although the bond strength remains the same.
| C-X bond | C-H | C-D |
| ~3000 | ~2200 |
Based on the equation, one would always expect a sharp lines at a well defined wavenumber. Unfortunately, the change in vibration modes is always accompanied with change in rotational mode (Stokes and Anti-Stokes). The required energy for this process is much smaller (1-5 cm-1) and causes together with some other effects the broading of the 'lines'.
The number of basic stretching and bending modes expected for a molecule increases with the number of atoms in the molecule. For non-linear molecules 3N-6 (2N-5 bending, N-1 stretching) vibrations are observed (e.g. CH2Cl2). For linear molecules e.g. CO2 one expect to find 3N-5 (3*3-5=4) modes. If there is no symmetry in the molecule most of them will be observed the IR spectrum; the remaining modes will be observed in a Raman spectrum. The more complicated the molecule is (the more atoms it possesses and the lower the symmetry), the more peaks can be observed in the IR spectrum (see example 3 and 4).
A signal is only observed in the IR spectrum, if the dipole momentum of the molecule changes during the interaction with the electromagnetic radiation. This is very likely for groups which already possess a significant dipole momentum to start with e.g. C-O, C-Cl, O-H, etc. (strong peaks). Groups with a small difference in electronegativity e.g. C-H, C-C, C=C, etc., will usually show weak or medium sized peaks in the IR spectrum.
An interpretation of an IR spectrum should include a detailed assignment of the peaks: exact wavenumber from the spectrum (integer), the intensity (w/m/s/br) and which functional group it represents, and maybe in addition the corresponding literature value. However, it is not necessary to interpret every little peak in the IR spectrum. Also, be cautious when you compare spectra which were obtain with different techniques (solution, Nujol mull, KBr pellet). The actual number of your vibration changes quite at bit, especially for highly polar compounds (Why ?).
2. Interpretation of the spectrum
The IR-spectrum can be divided into five ranges major ranges of interest for an organic chemist:
a. From 2700-4000 cm-1 (E-H-stretching: E=B, C, N, O)
In this range typically E-H-stretching modes are observed. The C-H-stretching modes can be found between 2850 and 3300 cm-1, depending on the hydrization. The range from 2850-3000 cm-1 belongs to saturated systems (alkanes, sp3, example 1), while the peaks from 3000-3100 cm-1 indicate an unsaturated system (alkenes, sp2, example 2; aromatic ring, example 3,4). Latter ones are usually weak or medium in intensity. The CH-function on a C-C-triple bond (alkynes) will appear as a sharp, strong peak around 3300 cm-1. The differences in wavenumbers are mainly due to different hybridization (=bond strength, the s-character in the bond, the stronger the bond is) and number of ligands on the carbon atom.
The O-H-stretching modes from alcohols and phenols (example 5) are mostly broad and very strong (3200-3650 cm-1) The O-H-peaks due to carboxylic acids (example 6) show a very broad and less intense peak between 2500 and 3500 cm-1. The change in peak shape is a result of the different degree of hydrogen bonds in alcohol and carboxylic acids. These peaks change significantly with the polarity of the solvent. They usually become sharper if the polarity of the solvent increases e.g. camphor in CCl4 and CH2Cl2 (see reader).
Peaks that are due to N-H-stretching modes are sharper than O-H-peaks (3300-3500 cm-1). Primary amines (example 7) have two peaks (sym./asym. vibration) in this range, while secondary amines (example 8) have only one peak. There are no peaks in this area for tertiary amines. Why?
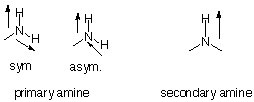
Two small or medium peaks at ~2750 and ~2850 cm-1 are a result of an aldehyde (example 15).
b. From 2000 - 2700 cm-1 (E-X-triple bonds: E=X=C, N, O)
This range covers mainly the triple bond stretching modes. The C-C-triple bond of alkynes (2130-2150 cm-1) is usually fairly weak, if observed at all. The C-N-triple bond of nitriles (example 10) (2100-2160 cm-1). In most cases a peak (with varying intensity) around 2349 cm-1 (together with 667 cm-1). This is due to the CO2 in the beam (poor background correction).
c. From 1500 - 2000 cm-1 (E-X-double bonds: E=X=C, N, O)
This is the most important range in the entire IR spectrum for organic chemists. If there is a very strong peak between 1640 and 1850 cm-1, there is most likely a carbonyl function in the molecule. Analysis of the exact peak position will reveal further what type of carbonyl function is present. The general rule is the more reactive the carbonyl compound is, the further to the right (=higher wavenumber) the C=O stretching frequency will be. The following sequence is observed:
acid chlorides > anhydrides > ester > aldehydes > ketones > carboxylic acids > amides
In order to identify a specific group additional information is needed from the other ranges of the IR spectrum:
| Carbonyl derivative | C=O-vibration (cm-1) | Example | Comment |
| acid chloride | 1780-1820 | example 11 | |
| carboxylic acid | 1700-1725 | example 6 | broad peak between 2500 and 3500 cm-1 |
| anhydride | ~ 1760 and ~1810 | example 12 | |
| ester | 1730-1750 | example 13 | medium or strong peak between 1000 and 1300 cm-1 |
| aldehyde | 1720-1740 | example 15 | weak or medium peaks peaks at 2750 and 2850 cm-1 |
| ketone | 1705-1725 | example 14 | |
| amide | 1630-1680 | example 16 | peaks at 3300-3500 cm-1 for primary or secondary amides |
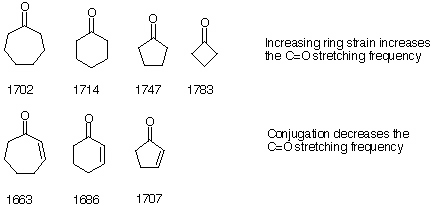
Ring strain usually increases the C=O stretching frequency. Conjugation of teh carbonyl group with other double bonds (aromatics, alkenes) or the formation of hydrogen bonds decreases the bond strength (shift: 15-60 cm-1 to lower wavenumbers) as the following examples of cyclic ketones demonstrate.
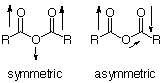
For all 'anhydride type systems' (O=C-X-C=O, X=O, NR, S, CH2) two peaks are observed in this region due to a symmetric and an asymmetric stretching mode.
In addition to the dominant C=O-mode, the C-C-double bond (example 2 -4 )is also located in this area (1600-1660 cm-1). However, if there is a carbonyl group present, it might be difficult to locate a weak or medium sized peak right next to it. Sometimes it is only observed as a shoulder. If there is a coupling between a C=C-group and other double bonded systems e.g. C=O or aromatic systems, the intensity will increase due to the increase in dipole momentum in the double bond. A medium or strong peak in this area corresponds to aromatic ring.
A nitro group shows two very intense peaks in the range between 1300-1400 cm-1 (sym.) and 1500-1600 cm-1 (asym.) (example 17).In a good spectrum, it might be possible to deduct the substitution pattern on an aromatic system from the overtone combination vibrations in the range from 1660-2000 cm-1 e.g. four peaks with increasing intensity are characteristic for a monosubstitution on your ring (example 4 and table for Arenes below).
d. From 1000-1500 cm-1 (E-X-single bonds, deformation, rocking modes)
A strong peak around 1450 cm-1 indicates the presence of methylene groups (CH2), while an additional strong peak about 1375 cm-1 is caused by a methyl group (CH3) (examples 1, 8-10). A symmetric 'doublet' with medium intensity around 1370 cm-1 is characteristic for an isopropyl group, while an asymmetric 'doublet' between 1365 and 1390 cm-1 is often due to a t-Bu-group.
The C-O-C-functions of ethers and esters are typically found as strong peaks in the range between 1000 and 1300 cm-1 (example 13). Generally, assignments in this area have to be done with extreme care, because there are a lot of ring absorbances in this ‘fingerprint area’.
e. Below 1000 cm-1 (out of plane modes, C-X: X=Cl, Br, I, heavier atoms)
This range belongs to the ’fingerprint area’, where assignments are a little bit uncertain. In some cases you can find information about the substitution pattern of alkenes or aromatic ring systems, if the range between 700-900 cm-1 is analyzed correctly (oop bending).
Alkenes
| Substitution pattern | Peaks at cm-1 | Intensity |
| monosubstitution | 895-915, 985-995 | strong, strong |
| cis-1,2-subst. | 680-730 | strong |
| trans-1,2-subst | 960-970 | strong |
| 1,1-disubst | 855-885 | strong |
| trisubst. | 790-830 | medium |
| tetrasubst. | ---------- | ---------- |
Arenes
| Substitution pattern | Peaks at cm-1 | Intensity |
| mono | 680-710, 720-760 | strong, strong |
| ortho (1,2) | 725-760 | strong |
| meta (1,3) | 670-710, 750-805, 870-900 | strong, strong, medium |
| para (1,4) | 800-860 | strong |
| 1,2,3 | 680-715, 750-780 | medium, strong |
| 1,3,5 | 670-700, 830-910 | medium, strong |
| 1,2,4 | 785-830, 870-900 | strong, medium |

Besides this information you may find strong C-Cl-vibrations around 740 cm-1 e.g. CH2Cl2.
3. Conclusion
Nobody is expected to memorize all those numbers. A lot of details can be deducted if a basic understanding of general trends is there. Although IR spectroscopy can reveal a lot of information about a molecule, but it is not enough to identify an unknown compound for sure. It is also important to keep in mind that an attempt to get too many details out of the ‘fingerprint area’ can be very misleading. In order to determine the nature of an unknown compound correctly, other techniques such as NMR (1H,13C, HETCOR, DEPT), Mass spectrometry, the chemical reactivity (solubility tests, sometimes derivatives) and physical properties (melting point, boiling point, refractive index) are required as well. Based on all these data, a structural suggestion for an unknown compound can be made.
4. Examples
Example 1: Alkane - Undecane
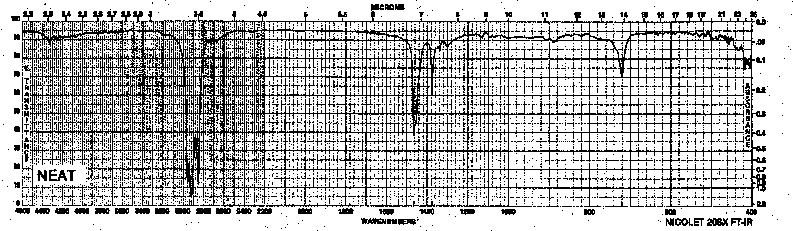
| Frequency in cm-1 | Assignment |
| 2924, 2850 | C-H (sp3, stretch) |
| 1467 | CH3 (sp3, bend) |
| 1378 | CH2, CH3 (sp3, bend) |
| 721 | for chain deformation (n>4) |
Example 2: Alkene - 1-Pentene
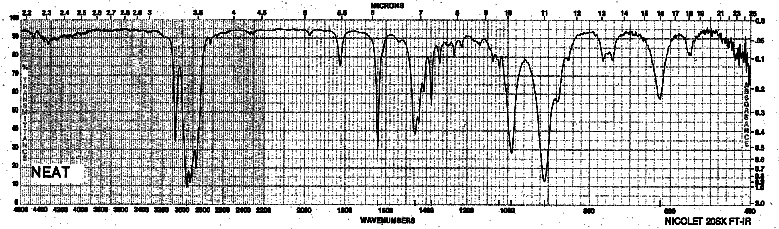
| Frequency in cm-1 | Assignment |
| 3080 | C-H (sp2, stretch) |
| 2965 | C-H (sp3, s)tretch |
| 1642 | C=C (stretch) |
| 992, 911 | =C-H (oop bending) |
Example 3: Arene - Benzene
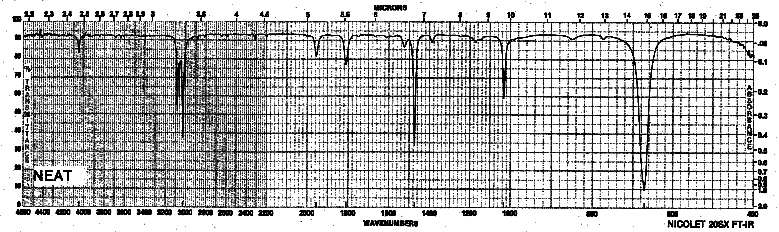
| Frequency in cm-1 | Assignment |
| 3035, 3060, 3090 | C-H (sp2, aromatic) |
| 1479 | C=C (stretch) |
| 1036 | =C-H (bending in ring plane) |
| 673 | =C-H (oop bending) |
Example 4: Substituted Arene - Ethylbenzene
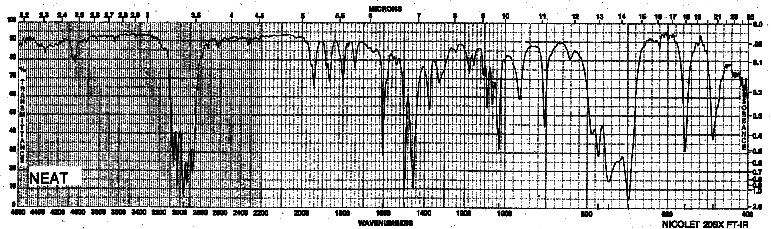
| Frequency in cm-1 | Assignment |
| 3030, 3050 | =C-H (sp2, stretch) |
| 2966 | C-H (sp3, stretch) |
| 1730-1940 | combination overtones (for a monosubstitution) |
| 1605, 1496 | C=C (stretch) |
| 1450 | CH2, CH3 (bend) |
| 746, 697 | C-H (oop bending, monosubst.) |
Example 5: Alcohols - Cyclohexanol
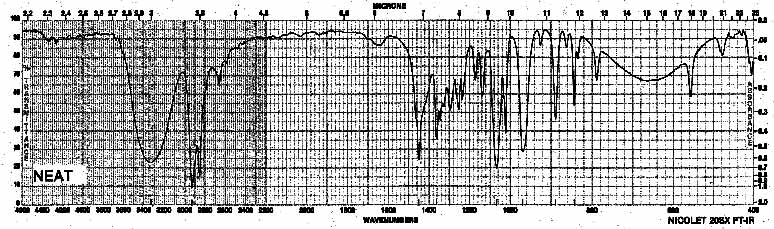
| Frequency in cm-1 | Assignment |
| 3600-3100 | O-H (stretch) |
| 2930, 2850 | CH2 (sp3, stretch) |
| 1067 | C-O (stretch) |
Example 6: Carboxylic Acid - Hexanoic Acid
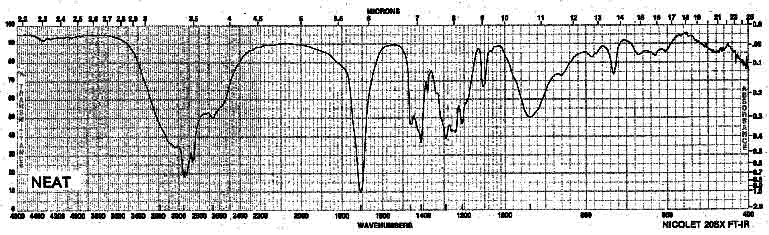
| Frequency in cm-1 | Assignment |
| 3500-2400 | O-H (stretch, O-H bridges) |
| 2932 | C-H (sp3, stretch) |
| 1711 | C=O (stretch) |
Example 7: Primary Amine - Propylamine
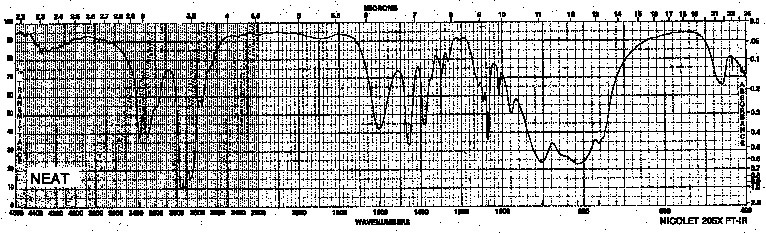
| Frequency in cm-1 | Assignment |
| 3367, 3280 | N-H (stretch, two peaks !) |
| 2958 | C-H (sp3, stretch) |
| 1607 | N-H (broad, scissoring) |
| 816 | N-H (broad, oop bending) |
Example 8: Secondary Amine - Diethylamine
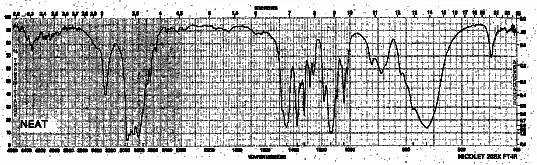
| Frequency in cm-1 | Assignment |
| 3280 | N-H (stretch, one peak only) |
| 2964 | C-H (sp3, stretch) |
| 1452 | CH3 (bend) |
| 1378 | CH2, CH3 (bend) |
| 776 | N-H (oop bending) |
Example 9: Tertiary Amine - Triethylamine
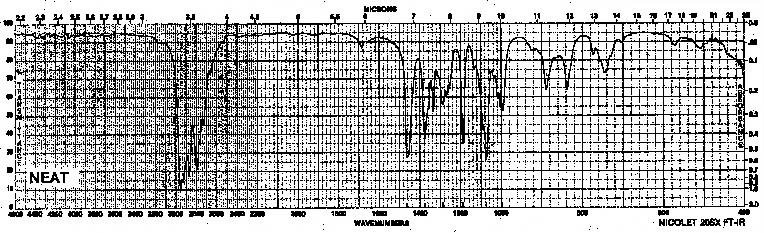
| Frequency in cm-1 | Assignment |
| 2980 | C-H (sp3, stretch) |
| 1460 | CH3 (stretch) |
| 1380 | CH2, CH3 (bend) |
Example 10: Nitrile - Butyronitrile
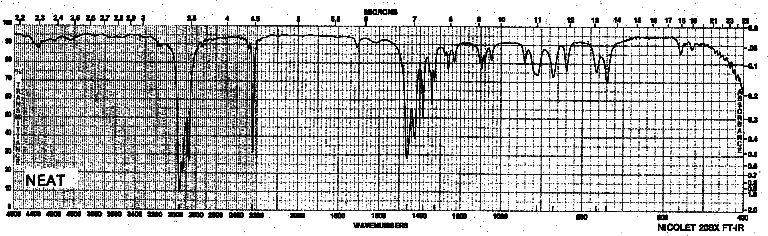
| Frequency in cm-1 | Assignment |
| 2973 | C-H (sp3, stretch) |
| 2250 | C=N (stretch) |
| 1465 | CH3 (stretch) |
| 1386 | CH2, CH3 (bend) |
Example 11: Carboxylic Acid Chloride - Valeryl chloride
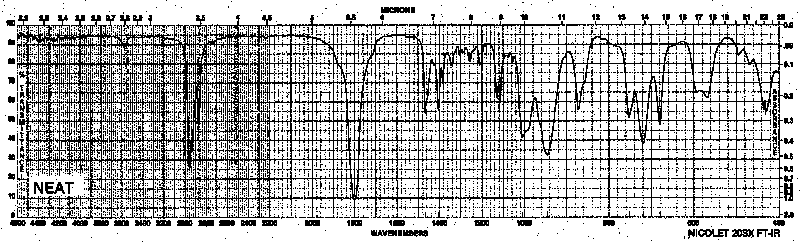
| Frequency in cm-1 | Assignment |
| 3560 | overtone from C=O peak |
| 2963 | C-H (sp3, stretch) |
| 1801 | C=O (stretch) |
| 753 | C-Cl (stretch) |
Example 12: Carboxylic Acid Anhydrides - Propionic anhydride
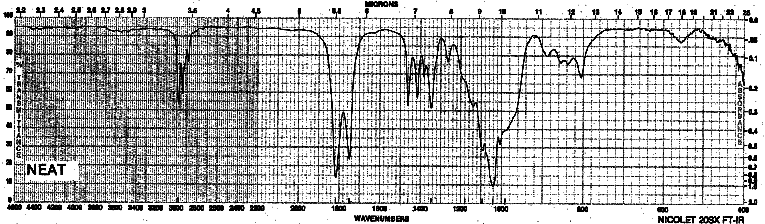
| Frequency in cm-1 | Assignment |
| 2987 | C-H (sp3, stretch) |
| 1819 | C=O (asym. stretch) |
| 1751 | C=O (sym. stretch) |
| 1094 | C-O (stretch) |
Example 13: Ester - Butyl acetate
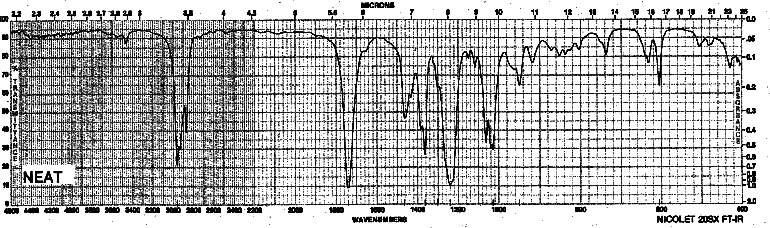
| Frequency in cm-1 | Assignment |
| 2962 | C-H (sp3, stretch) |
| 1743 | C=O (stretch) |
| 1243, 1031 | C-O-C (stretch) |
Example 14: Ketone - Cyclohexanone
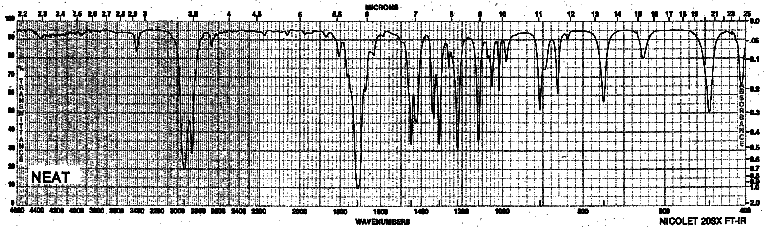
| Frequency in cm-1 | Assignment |
| 3405 | overtone from C=O peak |
| 2938 | C-H (sp3, stretch) |
| 1714 | C=O (stretch) |
Example 15: Aldehyde - Butyraldehyde
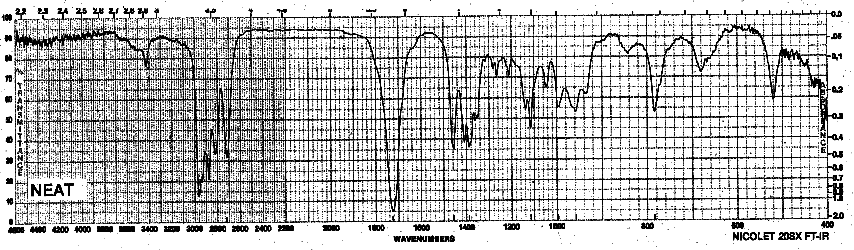
| Frequency in cm-1 | Assignment |
| 2950 | C-H (sp3, stretch) |
| 2820, 2720 | C-H (sp2, stretch, aldehyde) |
| 1720 | C=O (stretch) |
| 1460 | CH3 (stretch) |
| 1388 | CH2, CH3 (bend) |
Example 16: Amide - Propionamide
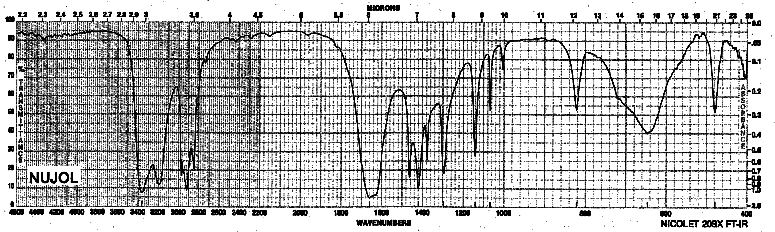
| Frequency in cm-1 | Assignment |
| 3362, 3200 | N-H (stretch, two peaks for a primary amide) |
| 2920 | C-H (sp3, stretch) |
| 1661 | C=O (stretch) |
Example 17: Nitro compounds - 1-Nitropropane
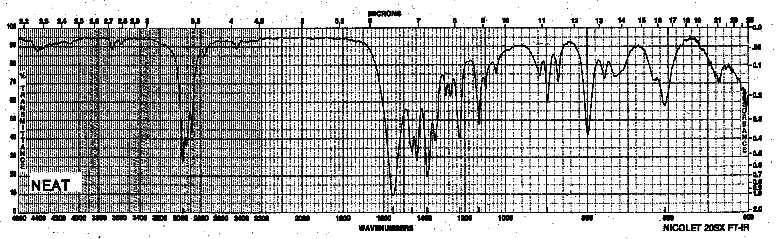
| Frequency in cm-1 | Assignment |
| 2978 | C-H (sp3, stretch) |
| 1554 | NO2 (asym. stretch) |
| 1387 | NO2 (sym. stretch) |