|
|
|
|
|
|
|
| Research |
 |
|
| The Houk group solves problems in organic and bio-organic chemistry using theoretical and computational methods
and programs. Some of the group members are also involved in experimental research to test theoretical predictions
and to develop new reactions, reagents, and catalysts which have been designed from theoretical investigations.
|
|
| Description of Research Subgroups |
 |
|
| INSIDE-OUT DESIGN OF NEW ENZYMES |
|
We are
involved in a collaboration
with David Baker of the University of Washington,
Department
of Biochemistry. This project involves a
completely general
and unique start-to-finish protocol for rapid
designing
novel enzyme catalysts for any desired chemical
reactions
using naturally occurring or de novo designed
protein
scaffolds.
We first build catalytic sites
unique catalytic site based upon chemical
precedent and
quantum mechanical calculations, followed by
construction
of a new stable protein that folds in such a way
so as
to produce the side-chain functionality
appropriately
oriented and in the proper environment to
constitute the
catalytic site. Recent publications in
Science (707) and Nature describe initial successes.
|
|
 |
|
UNDERSTANDING AND DESIGN OF STEREOSELECTIVE REACTIONS AND CATALYSTS |
|
We use computational methods to learn the origins of stereoselectivity in synthetically
useful reactions. This information, and quantitative calculations are used to design new reactions, reagents,
and catalysts. Our goals are to understand stereoselectivity, to make predictions, and to devise and execute
experimental tests of these predictions.
Christophe Allemann, Ruth Gordillo, Fernando R. Clemente, Paul Ha-Yeon Cheong, and K. N. Houk: "Theory of
Asymmetric Organocatalysis of Aldol and Related Reactions: Rationalizations and Predictions," Acc. Chem.
Res. 2004, 37, 558-569.
|
|
| PERICYCLIC REACTION MECHANISMS, RATES, AND DYNAMICS |
|
We use theory and computational methods to explore the transition states of pericyclic
reactions, some of the most significant reactions of organic chemistry. We explore the mechanisms of cycloadditions,
electrocyclizations, sigmatropic shifts, cheletropic reactions, and related processes. We are developing a general
theory of substituent effects on rates and stereoselectivities of these reactions.
Kelli S. Khuong, Chris M. Beaudry, Dirk Trauner, and K. N. Houk: "Dienophile Twisting and Substituent Effects
Influence Reaction Rates of Intramolecular Diels-Alder Cycloadditions: A DFT Study," J. Am. Chem. Soc.
2005, 127, 3688-3689.
|
|
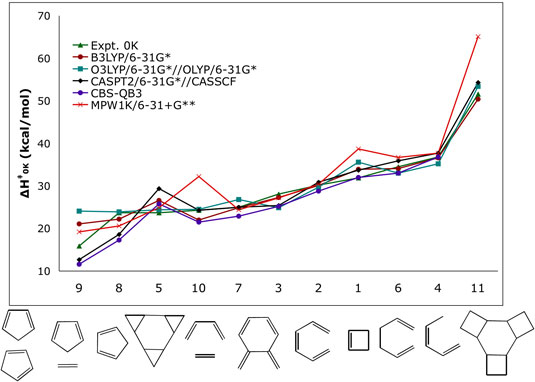
| BENCHMARKING COMPUTATIONAL METHODS FOR KINETICS AND
THERMODYNAMICS OF PERICYCLIC REACTIONS |
|
| Pericyclic reactions are utilized
in the syntheses of complex target molecules. The ability
to compute rates and equilibrium constants accurately
allows the prediction of the viability of proposed synthetic
routes and/or the design of successful alternatives. Current
high-accuracy computational methods can predict energies
within 1 kcal/mol of experiments. However, those calculations
require massive computational resources, and the system
size that can be treated is very limited and is usually
not applicable to real systems. The goal of this research
is to find optimal methodologies for the study of pericyclic
reactions. A variety of computational methods are being
used to study pericyclic reactions for which accurate
experimental data are available. This will allow us to
find methods that provide the same level of accuracy as
the most computationally expensive ones but that are also
fast enough to be of general and routine applicability
in pericyclic reactions. |
|
 |
|
| Figure 1. Computed activation enthalpies (kcal/mol) for a series of model pericyclic reactions. |
|
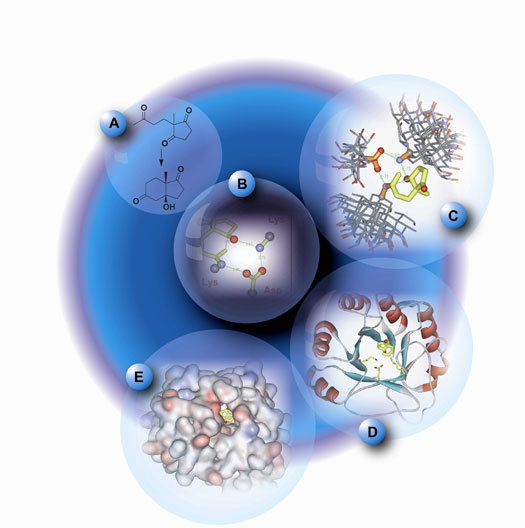
| EXPLORING THE ORIGINS OF ENZYME
CATALYSIS AND ANOMALOUS HIGH AFFINITY BINDERS |
|
Enzymes are exceptional catalysts
with average 22 kcal/mol and maximum 38 kcal/mol binding
energies toward transition states.1 Such strong binding
could not be accounted for solely by noncovalent interactions
proposed by Pauling and others, such as hydrogen bonds,
electrostatics, van der Waals, etc. Those factors can
contribute only up to 15 kcal/mol for binding. We
have proposed
that most enzymes achieve over 1011 M-1 proficiency by
full or partial covalent bond formation to the reacting
substrate in the transition state, involving a change
in chemical mechanism from that occurring in the uncatalyzed
reaction in aqueous solution. Intermediates covalently
bound to the enzyme or cofactor, proton transfer (general
acid/base catalysis and low-barrier hydrogen bonds) occurring
in the transition state, and bonding to metal cations
in the transition state all fall into the category of
covalent catalysis.2
Using our insights about covalent catalysis, we explore
the origins of proficient enzyme catalysis and selectivity,
including ODCase, aldolase, lipase, protease, NO synthase,
etc. by modeling theozymes - an array of functional
groups necessary for catalysis3 - with density functional
theory. The goal is to understand how covalent and noncovalent
interactions provide catalysis at a molecular level.
We also examine the ultra scale binding of biotin and
femtomolar antibodies with theozyme modeling in order
to rationalize how noncovalent binding greater than
15 kcal/mol is feasible in these few examples.
|
|
 |
|
1. Houk, K. N.; Leach, A. G.; Kim, S. P.; Zhang, X.:
"Binding Affinities of Host-Guest, Protein-Ligand,
and Protein-Transition-State Complexes,"
Angew. Chem. Int. Ed. 2003, 42, 4872-4897.
2. Zhang, X.: Houk, K. N.: "Why Enzymes Are Proficient
Catalysts: Beyond the Pauling Paradigm," Acc.
Chem. Res. 2005, ASAP.
3. Tantillo, D. J.; Chen, J.; Houk, K. N.: "Theozyme
and Compuzymes: Theoretical Models for Biological Catalysis,"
Curr. Opin. Chem. Biol. 1998, 2, 743-750.
|
|
| REACTIVE INTERMEDIATES IN BIOLOGY AND THE ATMOSPHERE |
|
We use theoretical methods to explore the reactions undergone by neurotransmitters
like NO and the functional equivalents such as RSNOs and NO precursors under various conditions. We learn
how nitrogen and sulfur oxides react in biological systems and the atmosphere. We explore the roles of
dynamics, entropy, solvation, and tunneling on the very rapid reactions of excited states, carbenes, singlet
oxygen, radicals, and other reactive intermediates.
Michael D. Bartberger, Wei Liu, Eleonora Ford, Katrina M. Miranda, Christopher Switzer, Jon M. Fukuto,
Patrick J. Farmer, David A. Wink, and Kendall N. Houk: "The Reduction of Potential of Nitric Oxide (NO) and its
Importance to NO Biochemistry," Proc. Natl. Acad. Sci. USA 2002, 99, 10958-10963.
|
|
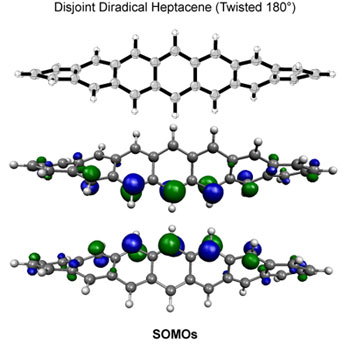
| STRUCTURES AND PROPERTIES OF ORGANIC MATERIALS |
|
|
|
|
 |
|
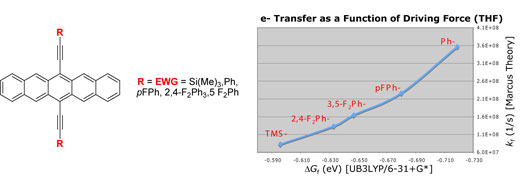
| Polycyclic
aromatic compounds have shown great promise for use in
organic electronic materials. In particular, thin film
transistors of pentacene have been shown to exhibit charge
carrier mobilities comparable to silicon. However, marketable
devices based on pentacene have not been realized due
to the ease with which pentacene is photooxidized. The
mechanism of photooxidation is believed to proceed via
photostimulated electron transfer to oxygen, leading to
other products and the loss of any desirable electronic
properties, although photooxidation via energy transfer
to form singlet oxygen is also reasonable.5 A greater
understanding of the mechanism may lead to the development
of substituted pentacenes that overcome the stability
problem. |
|
|
|
1. Norton, J. E.; Houk, K. N. J. Am. Chem. Soc. 2005, 127,
4162-4163.
2. Lu, J.; Ho, D. M.; Vogelaar, N. J.; Kraml, C. M.; Pascal, R. A., Jr. J. Am. Chem. Soc. 2004,
126, 11168-11169.
3. Houk, K. N.; Lee, P. S.; Nendel, M. J. Org. Chem. 2001, 66, 5517-5521.
4. Bendikov, M.; Duong, H. M.; Starkey, K.; Houk, K. N.; Carter, E. A.; Wudl, F. J. Am. Chem. Soc. 2004, 126, 7416-7417.
5. Maliakal, A.; Raghavachari, K.; Katz, H.; Chandross, E.; Siegrist, T. Chem Mater. 2004, 16, 4980-4986.
6.
Anthony, J. E.; Brooks,J. S.; Eaton, D. L.; Parkin, S. R. J. Am. Chem. Soc. 2001, 123, 9482-9483.
|
|
| GATING IN HOST-GUEST COMPLEXES |
|
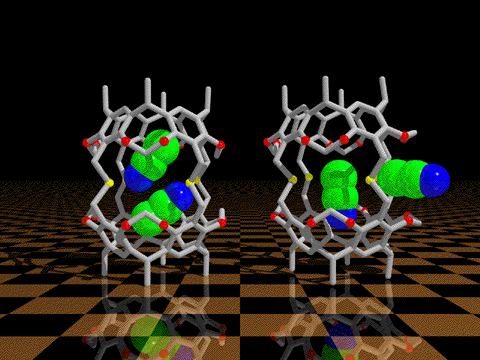
| We have discovered that conformational processes of host molecules can control
the stabilities of host-guest complexes. We use theory to design gated cavitands that have a movable
lid that will bind a wide range of small guest molecules, and release them only upon gate opening by a
conformational change induced by heat, photochemistry, or redox processes. We undertake the synthesis
and experimental studies of such controlled release container molecules.
|
|
|
|
|
K. N. Houk, Kensuke Nakamura, Chimin Sheu, and Amy E. Keating: "Gating as a Control Element in
Constrictive Binding and Guest Release by Hemicarcerands," Science 1996, 273, 627-629.
|
|
|
|
|