 We have fully characterized the spectroscopy of the Na¯/THF
system, which now represents the only electron transfer reaction for
which the spectrum of the reactant, its excited state, and both products
are fully known and can be monitored independently as the reaction
progresses. What we found is that photoexcitation of the
sodium anion creates a localized excited state which is bound by the
polarization of the solvent surrounding the anion. As the solvent
rearranges in response to this excitation, the electron is ejected from
the sodium nucleus into a nearby solvent cavity. In THF, the
electron transfer takes ~700 fs, a time scale that suggests that solvent
translational motions are responsible for the charge transfer dynamics;
this result is consistent with computer simulations also being performed
by our group. By comparing the behavior of the solvated
electrons produced via charge transfer to those created by direct
ionization of the solvent, we have inferred many of the molecular
details of electron transfer reactions, including: the distance
electrons are ejected following excitation, the stability of the
resulting atom:electron contact pairs, and the fact that back electron
transfer occurs slowly because it is in the Marcus inverted
regime. Our basic picture summarizing the photochemistry of the
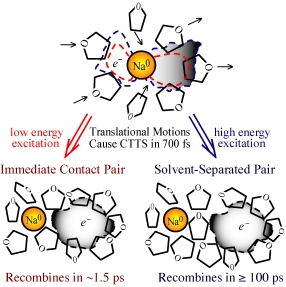
sodium anion is shown at left: the initial absorption of light
takes the extra 3s electron on the Na¯ and promotes it to a p-like
excited state bound by the surrounding solvent. As the
first-shell molecules translate in response to this excitation, they
"squeeze" some of the electron density and force the electron off the Na
atom. For low energy excitation (red-dashed wavefunction), the
spatial extent of the wavefunction is small and the electron is ejected
adjacent to the Na atom. Since the ejected electron's wavefunction
can still overlap with the Na nucleus, recombination occurs quickly, in
under 2 picoseconds. For higher energy excitation (blue-dashed
wavefunction), however, the wavefunction has a much greater curvature,
including significant probability density beyond the first solvent
shell. Thus, when the electron is subsequently ejected, there is
a large probability that it ends up one solvent shell away from the Na
atom. This "solvent-separated" contact pair is a relatively
stable entity: the electron and Na atom cannot simply diffuse
apart because it would cost a great deal of free energy to build
entirely new solvent shells around each species. It is also
difficult for the electron to go back onto the Na atom for the same
reason: it costs energy to disrupt the solvent structure around
the separated pair. For THF at room temperature, a fluctuation
that disrupts the local structure occurs on average once every few
hundred picoseconds, leading to very slow recombination of the electron
with its geminate Na atom partner. The same solvent motions are
responsible for the electron ejection process no matter what the
excitation energy, so the 700 fs time scale for charge ejection is
independent of excitation wavelength. This picture is consistent
with 3-pulse experiments in which we control the distance between the
electron and the sodium atom by directly exciting the electron at
different times after it has been ejected. When electrons in
immediate contact pairs are excited within the first 1.5 ps after
ejection, the delocalization of the electronic wavefunction upon
excitation produces an increased probability of finding the electron
far from its Na atom partner, leading to a decrease in amount of
back electron transfer (recombination). When the electrons
in solvent separated contact pairs that exist at longer times (> 4
ps) after ejection are excited, however, the delocalization of the
electronic wavefunction increases the probability of finding the
electron in the same solvent cavity as the Na atom, enhancing the
amount of recombination. Finally, we have completed a study of the
electron photodetachment process of Na¯ in a variety of different
solvents, and we have been able to extract the position of the
"conduction band" out from under the Na¯ absorption
spectrum. See, e.g.,
We have fully characterized the spectroscopy of the Na¯/THF
system, which now represents the only electron transfer reaction for
which the spectrum of the reactant, its excited state, and both products
are fully known and can be monitored independently as the reaction
progresses. What we found is that photoexcitation of the
sodium anion creates a localized excited state which is bound by the
polarization of the solvent surrounding the anion. As the solvent
rearranges in response to this excitation, the electron is ejected from
the sodium nucleus into a nearby solvent cavity. In THF, the
electron transfer takes ~700 fs, a time scale that suggests that solvent
translational motions are responsible for the charge transfer dynamics;
this result is consistent with computer simulations also being performed
by our group. By comparing the behavior of the solvated
electrons produced via charge transfer to those created by direct
ionization of the solvent, we have inferred many of the molecular
details of electron transfer reactions, including: the distance
electrons are ejected following excitation, the stability of the
resulting atom:electron contact pairs, and the fact that back electron
transfer occurs slowly because it is in the Marcus inverted
regime. Our basic picture summarizing the photochemistry of the
sodium anion is shown at left: the initial absorption of light
takes the extra 3s electron on the Na¯ and promotes it to a p-like
excited state bound by the surrounding solvent. As the
first-shell molecules translate in response to this excitation, they
"squeeze" some of the electron density and force the electron off the Na
atom. For low energy excitation (red-dashed wavefunction), the
spatial extent of the wavefunction is small and the electron is ejected
adjacent to the Na atom. Since the ejected electron's wavefunction
can still overlap with the Na nucleus, recombination occurs quickly, in
under 2 picoseconds. For higher energy excitation (blue-dashed
wavefunction), however, the wavefunction has a much greater curvature,
including significant probability density beyond the first solvent
shell. Thus, when the electron is subsequently ejected, there is
a large probability that it ends up one solvent shell away from the Na
atom. This "solvent-separated" contact pair is a relatively
stable entity: the electron and Na atom cannot simply diffuse
apart because it would cost a great deal of free energy to build
entirely new solvent shells around each species. It is also
difficult for the electron to go back onto the Na atom for the same
reason: it costs energy to disrupt the solvent structure around
the separated pair. For THF at room temperature, a fluctuation
that disrupts the local structure occurs on average once every few
hundred picoseconds, leading to very slow recombination of the electron
with its geminate Na atom partner. The same solvent motions are
responsible for the electron ejection process no matter what the
excitation energy, so the 700 fs time scale for charge ejection is
independent of excitation wavelength. This picture is consistent
with 3-pulse experiments in which we control the distance between the
electron and the sodium atom by directly exciting the electron at
different times after it has been ejected. When electrons in
immediate contact pairs are excited within the first 1.5 ps after
ejection, the delocalization of the electronic wavefunction upon
excitation produces an increased probability of finding the electron
far from its Na atom partner, leading to a decrease in amount of
back electron transfer (recombination). When the electrons
in solvent separated contact pairs that exist at longer times (> 4
ps) after ejection are excited, however, the delocalization of the
electronic wavefunction increases the probability of finding the
electron in the same solvent cavity as the Na atom, enhancing the
amount of recombination. Finally, we have completed a study of the
electron photodetachment process of Na¯ in a variety of different
solvents, and we have been able to extract the position of the
"conduction band" out from under the Na¯ absorption
spectrum. See, e.g., I. B. Martini and B. J. Schwartz, "Elucidating
the Initial Dynamics of Electron Photodetachment from Atoms in Liquids
Using Variably-Time-Delayed Resonant Multiphoton Ionization," J. Chem. Phys. 121(1), 374-9 (2004).
E. R. Barthel and B. J. Schwartz, "Mapping the Conduction Band Under CTTS Transitions: The Photodetachment Quantum Yield of Sodide (Na¯) in Tetrahydrofuran," Chem. Phys. Lett. 375(3-4), 435-43 (2003).
E. R. Barthel, I. B. Martini, E. Keszei and B. J. Schwartz, "Solvent
Effects on the Ultrafast Dynamics and Spectroscopy of the
Charge-Transfer-to-Solvent (CTTS) Reaction of Sodide," J. Chem.
Phys. 118(13), 5916-31 (2003).
I. B. Martini, E. R. Barthel and B. J. Schwartz, "Manipulating the Production and Recombination of Electrons During Electron Transfer: Femtosecond Control of the Charge-Transfer-to-Solvent (CTTS) Dynamics of the Sodium Anion," J. Am. Chem. Soc. 124(25), 7622-34 (2002).
I. B. Martini and B. J. Schwartz, "On the Insensitivity of the Non-Adiabatic Relaxation of Solvated Electrons to the Details of their Local Solvent Environment," Chem. Phys. Lett. 360(1-2), 22-30 (2002).
I. B. Martini, E. R. Barthel and B. J. Schwartz, "Optical Control of Electrons During Electron Transfer," Science 293, 462-5 (2001).
E. R. Barthel, I. B. Martini and B. J. Schwartz, "How Does the Solvent Control Electron Transfer? Experimental and Theoretical Studies of the Simplest Charge Transfer Reaction," FEATURE ARTICLE, J. Phys. Chem. B 105(49), 12230-41 (2001).
I. B. Martini, E. R. Barthel and B. J. Schwartz, "Mechanisms of the Ultrafast Production and Recombination of Solvated Electrons in Weakly-Polar Fluids: Comparison of Multiphoton Ionization and Detachment via the Charge-Transfer-to-Solvent Transition of Na¯ in THF," J. Chem. Phys. 113(24), 11245-57 (2000).
E. R. Barthel, I. Martini and B. J. Schwartz, "Direct
Observation of Charge-Transfer-to-Solvent (CTTS) Transitions: Ultrafast
Dynamics of the Photoexcited Alkali Metal Anion Sodide (Na¯)," J.
Chem. Phys. 112(21), 9433-44 (2000).
For others' perspective on our work in this area, follow some of
the links on the Schwartz
Group Press page.
This work was supported in part by the National
Science Foundation under CAREER award CHE-9733218