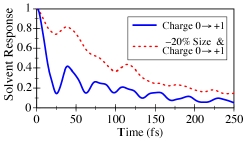
 a chemical reaction. In much of the previous work in this area,
the solvent relaxation was studied by suddenly changing the solute's
charge without changing its size (e.g., the blue solid curve in the
Figure). For this case, over 80% of the solvent relaxation occurs
in the first 25 femtoseconds. A detailed analysis shows that it
is water librational motions (i.e. hindered rotations that lead to
reorientation of the solvent dipoles) that cause this rapid
relaxation. Real solutes, however, undergo changes in size
and shape as well as charge during a chemical reaction: after
all, removing or adding an electron to a solute certainly changes its
size! When this more realistic type of combined solute
size/charge change is simulated (red dashed curve), the solvent
response is nearly an order of magnitude slower than for the case for
changing only the charge: it takes 200 femtoseconds to complete
80% of the relaxation. The analysis shows that most of the
solvation energy is carried by the relatively slow translational
motions of just the closest one or two solvent molecules. Even in
a polar solvent like water, a size change of only 10-15% is enough to
cause translational motions to dominate, whether or not there is also
an accompanying charge change. Since most reactions involve some
change in size, we believe that "standard" theories in which the
solvent is treated as a dielectric continuum are not the best way to
approach the problem of solvent effects on chemical reactivity;
instead, we feel that the dynamics of electron transfer and many other
solvent-controlled reactions are dominated by the translational motions
of just one or two nearby solvent molecules.
a chemical reaction. In much of the previous work in this area,
the solvent relaxation was studied by suddenly changing the solute's
charge without changing its size (e.g., the blue solid curve in the
Figure). For this case, over 80% of the solvent relaxation occurs
in the first 25 femtoseconds. A detailed analysis shows that it
is water librational motions (i.e. hindered rotations that lead to
reorientation of the solvent dipoles) that cause this rapid
relaxation. Real solutes, however, undergo changes in size
and shape as well as charge during a chemical reaction: after
all, removing or adding an electron to a solute certainly changes its
size! When this more realistic type of combined solute
size/charge change is simulated (red dashed curve), the solvent
response is nearly an order of magnitude slower than for the case for
changing only the charge: it takes 200 femtoseconds to complete
80% of the relaxation. The analysis shows that most of the
solvation energy is carried by the relatively slow translational
motions of just the closest one or two solvent molecules. Even in
a polar solvent like water, a size change of only 10-15% is enough to
cause translational motions to dominate, whether or not there is also
an accompanying charge change. Since most reactions involve some
change in size, we believe that "standard" theories in which the
solvent is treated as a dielectric continuum are not the best way to
approach the problem of solvent effects on chemical reactivity;
instead, we feel that the dynamics of electron transfer and many other
solvent-controlled reactions are dominated by the translational motions
of just one or two nearby solvent molecules. We also have developed a new formalism specifically designed to pick out the individual motions of solvent molecules that are important in relaxation. Using this formalism, we have found that even when the solvent relaxation for different perturbations takes place on the same time scale, the underlying solvent motions can still be completely different. This indicates that the theory of linear response, one of the cornerstones of nonequilibrium statistical mechanics, fails for many systems of interest, and that the failure is often hidden by a coincidental similarity of the relaxation time for different solvent motions. See, e.g.,
M. J. Bedard-Hearn, R. E. Larsen and B. J. Schwartz, "Understanding Nonequilibrium Solvent Motions Through Molecular Projections: Computer Simulations of Solvation Dynamics in Liquid Tetrahydrofuran (THF)," J. Phys. Chem. B 107(51), 14464-75 (2003).
M. J. Bedard-Hearn, R. E. Larsen and B. J. Schwartz, "Hidden Breakdown of Linear Response: Projections of Molecular Motions in Nonequilibrium Simulations of Solvation Dynamics," J. Phys. Chem. A 107(24), 4773-7 (2003).
D. Aherne, V. Tran and B. J. Schwartz, "Non-Linear, Non-Polar Solvation Dynamics in Water: The Roles of Electrostriction and Solvent Translation in the Breakdown of Linear Response," J. Phys. Chem. B 104(22), 5382-94 (2000).
V. Tran and B. J.
Schwartz, "The
Role of Non-Polar Forces in Aqueous Solvation: Computer Simulation
Study of Solvation Dynamics in Water Following Changes in Solute Size,
Shape and Charge," J. Phys. Chem. B 103 (26), 5570-80
(1999).
We also have
evidence for the importance of solvent translational motions in
controlling electron transfer dynamics in our
experiemental work on the sodium anion.
This
work was supported in part by the National Science Foundation under
CAREER award CHE-9733218 and grant CHE-0240776.