|
|
|
|
|
|
|
|
|
|
|
| Individual Research Projects |
 |
|
| Potential Energy Surfaces for Diradical-Mediated
Rearrangements and the Role of Dynamics |
 |
|
Christopher P. Suhrada and
K. N. Houk
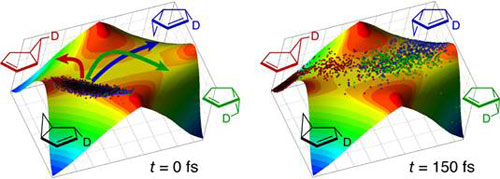
Bicyclo[3.1.0]hex-2-ene.
Labeled bicyclo[3.1.0]hex-2-ene rearranges to degenerate
products in unequal proportions, despite the intermediacy
of a single, symmetrical diradical structure. We have
mapped the PES and concluded that the central diradical,
because it has essentially no energy minimum, behaves
more as a transition state than as an intermediate. Accordingly,
the three products are formed by equi-energetic but geometrically
non-equivalent paths. Based on our data, Charles Doubleday
(Columbia University) performed molecular dynamics simulations,
illustrated below, which show how the PES shape influences
reaction trajectories to produce the experimentally observed
distribution. |
|
 |
|
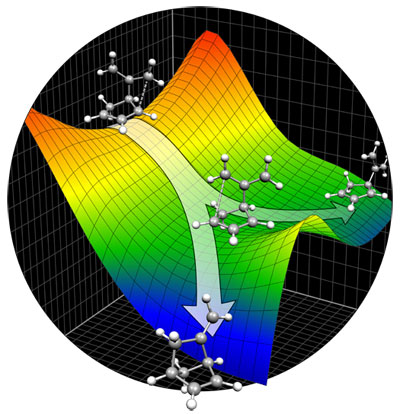
6-Methylenebicyclo[3.2.0]hept-2-ene.
Experiments by Dieter Hasselmanns group (Ruhr-Universität
Bochum) demonstrated that labeled 6-methylenebicyclo[3.2.0]
hept-2-ene rearranges to 5-methylenenorbornene with partial
regio- and steroselectivity. Our theoretical findings
attribute much of the observed selectivity to a stepwise
mechanism with multiple, non-degenerate paths from reactant
to intermediates and from intermediates to products,
as well as incomplete conformational equilibration of
diradicals. A fit to the experimental data implicates
a dynamical bifurcation of some bond-breaking trajectories
toward either to the intermediate, on one hand, or directly
to the product, on the other (illustrated below). |
|
 |
|
| Molybdenum-Catalyzed Asymmetric Allylations |
|
|
J. A. R. Luft and
K. N. Houk
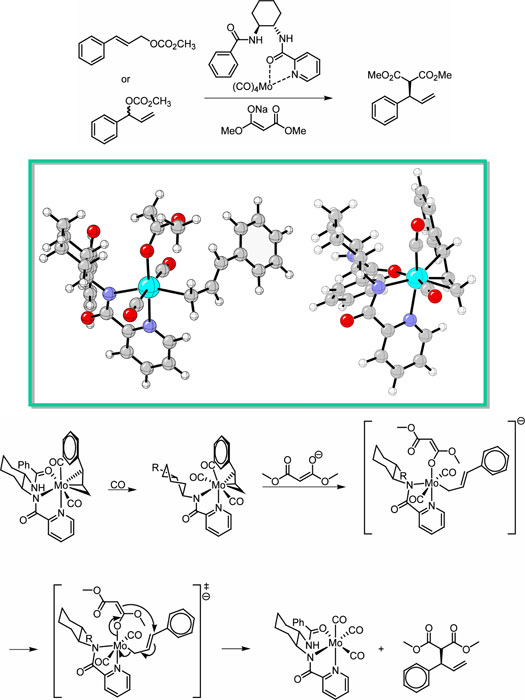
Molybdenum-catalyzed asymmetric allylations have become
a powerful method to afford a branched substrate in high
yield and enantiomeric excess from either the linear or
branched reagent. Shane Krska and his co-workers at Merck
Research Laboratories have been working to understand
the mechanism of this unusual reaction. The reaction is
known to proceed via a π
-allyl complex. The reaction requires an external source
of CO. Previous studies have shown that both oxidative
addition and nucleophilic attack occur with retention
of configuration. Computational studies using Hartree-Fock
and Density Functional Theory have begun on the known
and anticipated intermediates. Preliminary calculations
led to the potential reaction mechanism of calculated
intermediates are shown. |
|
 |
|
| Krska, S. W.; Hughes, D. L.; Reamer,
R. A.; Mathre, D. J.; Sun, Y. The Unusual Role of CO
Transfer in Molybdenum-Catalyzed Asymmetric Alklyations
J. Am. Chem. Soc. 2002, 124, 12656-12657. |
|
| Intramolecular Halocyclizations |
|
|
J. A. R. Luft and
K. N. Houk
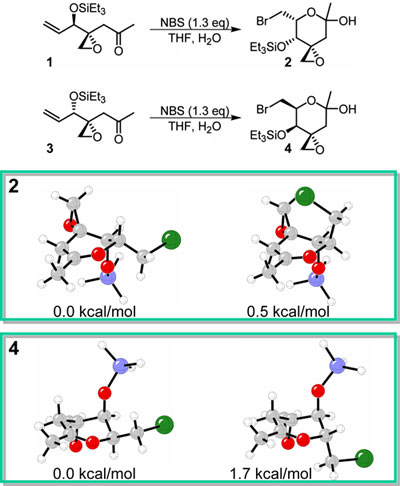
Intramolecular halocyclizations have been previously studied
both experimentally and theoretically. We are currently
investigating a highly diastereoselective bromolactolization.
Kazunori Koide at the University of Pittsburgh reasoned
that reacting 1 with NBS in water would
afford a diastereomeric mixture of bromolactol. He planned
to separate the desired stereoisomer and proceed with
his synthesis of FR901464.1 Unexpectedly, the cis isomer
was formed in high yield (75%). A substrate with opposite
stereochemistry at the siloxy substituent, 3,
afforded 4 with even higher selectivity
(95%). Computational studies show that the strong conformational
preference of the siloxy group determines the lowest energy
conformers. The conformational preferences of the remaining
substituents affect the relative stabilities of the cis
and trans isomers. These stabilities explain the observed
stereoselectivities. |
|
 |
|
| 1. Albert, B. J.;
Koide, K. Synthesis of a C4-epi-C1-C6 Fragment of FR901464
Using a Novel Bromolactolization, Org. Lett.
2004, 6, 3655-3658. |
|
| An Unexpected Radical Rearrangement En Route to Avermectin B1 |
|
|
J. A. R. Luft and
K. N. Houk
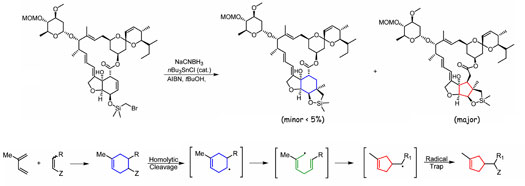
During the synthesis of an Avermectin B1 analogue, chemists
observed an unexpected radical rearrangement under Nishiyama
conditions for radical ring formation. The expected product,
containing a cyclohexyl
ring, was observed in less than 5%. The major product
was a five-five-five
fused ring system from an apparent radical ring rearrangement.
Computational methods have been used to explain the cause
of the rearrangement. Computational efforts indicate that
the macrocycle is not necessary to afford the rearrangement.
Experimental investigations are currently focused on radical
reactions that might afford the rearranged products. One
proposed reaction would yield a substituted cyclopentyl
ring in two to three steps starting from a Diels-Alder
reaction of substrates of interest. |
|
 |
|
| Nickel Coupling Mechanism |
|
|
Patrick R. McCarren and
K. N. Houk
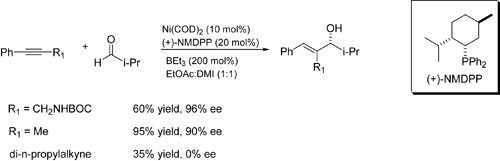
The intermolecular, stereoselective reaction below may have great utility.
|
|
 |
|
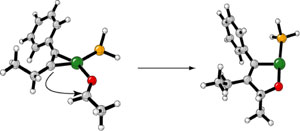
| How does it work? Computational chemistry
is testing proposed mechanisms such as the stereocenter-forming
step shown below. Our goal is to gain mechanistic insight
and a predictive model for future experiments. |
|
 |
|
| Jamison et al. J. Am. Chem. Soc.
2003, 125, 3442-3443. |
|
| Understanding the Proficiency of
OMP Decarboxyzlase: An Ab Initio Study |
|
|
Courtney L. Stanton and
K. N. Houk
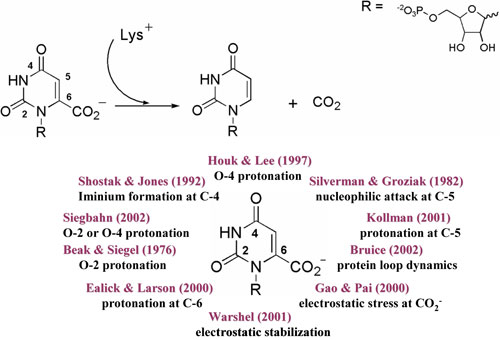
Orotidine 5'-Monophosphate Decarboxylase.
In 1995 the enzyme orotidine 5'-monophosphate decarboxylase
(OMP decarboxylase) was declared the most proficient*
enzyme known (Radzicka, A.; Wolfenden, R. Science
1995, 267, 90-92). Despite a
decade of attention and the proposal of many different
hypotheses, the mechanism of ODCase catalysis remains
unsolved (for review see Houk, K. N.; Tantillo, D.J.;
Stanton, C.; Hu, Y. Topics in Current Chemistry
2004, 238, 1-22).
We have undertaken a theoretical exploration of all relevant
reaction mechanisms that have been suggested for OMP decarboxylase
by applying density functional methods to small model
systems. Our intention is to gain a systematic knowledge
of the inherit advantage that one type of catalysis may
have over another. |
|
|
|
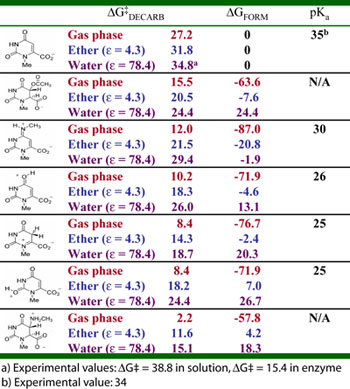
Density Functional Calculations
Table 1. Reaction energetics (kcal/mol)
for the decarboxylation of various N-methyl orotic acid
derivatives. All calculations done at the B3LYP/6-31+G(d,p)
level, using CPCM implicit solvent model with UAKS radii
where applicable. ΔGFORM
refers to formation of the pictured intermediates from
N-methyl orotic acid. pKa values refer to the carbon acidity
at C6 for each decarboxylated intermediate and are calculated
at the same level of theory and implicit solvent model. |
|
 |
|
Characterization of Synthetic Linear
Motor Molecule Actuation
via Atomic Force Spectroscopy and Computational Modeling |
|
|
Brian H. Northrop, K. N.
Houk, J. Fraser Stoddart, B. Brough, J. J. Schmidt, H.-R.
Tseng, and C.-H. Ho
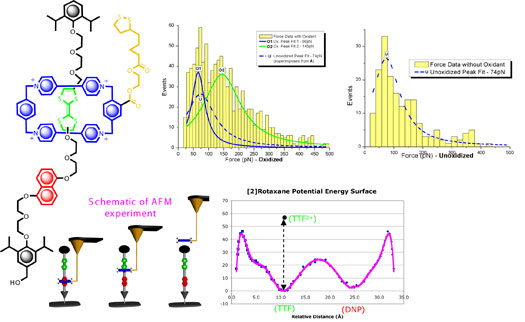
Molecular motors have recently garnered great interest
for their potential to convert energy into mechanical
work at the micro and nanoscale. Being able to quantitatively
measure the amount of energy available to for a molecular
scale motor do work is necessary for the characterization,
development, and optimization of molecular motors. To
this aim, an interdisciplinary combination of synthesis,
surface chemistry, single molecule atomic force microscopy
(AFM), and theoretical molecular dynamics simulations
have been used to characterize the actuation of a synthetic
[2]rotaxane-based motor molecule. The specifically designed
[2]rotaxane has been synthesized with a dual recognition
motif to allow for attachment to both a silicon surface
and a gold AFM tip. Molecular dynamics and mechanics calculations
were used to map the potential energy surface of the [2]rotaxane
and, in conjunction with atomic force measurements, have
been used to provide a quantitative description of molecular
actuation. These results were further supported by density
functional calculations. |
|
 |
|
| Brough, B.; Northrop, B. H.; Schmidt,
J. J.; Tseng, H.-R., Houk, K. N.; Stoddart, J. F.; Ho,
C.-H. submitted. |
|
| Pentacene Physical Properties and
Photostability, a Theoretical and Experimental Mechanistic
Study |
|
|
Brian H. Northrop, K. N.
Houk, J. Fraser Stoddart, and Elsa Reichmanis
Polycyclic aromatic compounds have shown great promise
for use in organic electronic materials. In particular,
thin film transistors of pentacene have been shown to
exhibit charge carrier mobilities comparable to silicon.
However, marketable devices based on pentacene have not
been realized due to the ease with which pentacene is
photooxidized. The mechanism of photooxidation is believed
to proceed via photostimuated electron transfer to oxygen,
leading to other products and the loss of any desirable
electronic properties, although photooxidation via energy
transfer to form singlet oxygen is also reasonable. A
greater understanding of the mechanism may lead to the
development of substituted pentacenes that overcome the
stability problem.
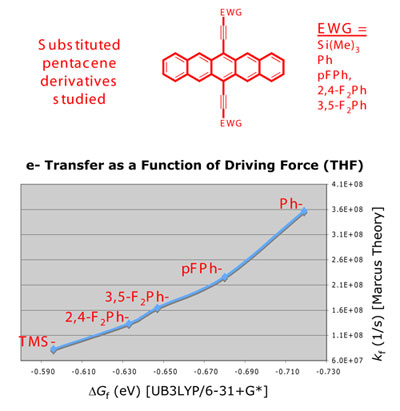
A combination of synthesis, photostability measurements,
and theoretical calculations is being used to determine
whether pentacene photooxidation occurs via electron or
energy transfer. Building upon the work of Anthony, a
series of 6,13-disubstituted pentacene derivatives have
been synthesized. Photostability measurements are currently
being performed in tetrahydrofuran and toluene. Additionally,
density functional calculations are being used to predict
rates of electron transfer using Marcus theory:
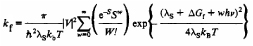
The results should establish the mechanism of photooxidaion
and lead to more stable pentacenes.
|
|
 |
|
| Maliakal, A.; Raghavachari, K.;
Katz, H.; Chandross, E.; Siegrist, T. Chem Mater.
2004, 16, 4980-4986. Anthony,
J. E.; Brooks,J. S.; Eaton, D. L.; Parkin, S. R. J.
Am. Chem. Soc. 2001, 123,
9482-9483. |
|
| Theorectical Studies of Quantum
Amplification of Isomerization for Imaging Systems |
|
|
| Joseph E. Norton, Leif
P. Olson, and K. N. Houk
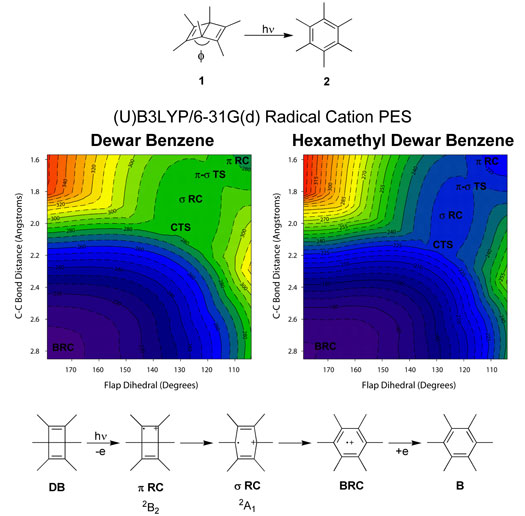
Quantum Amplified Isomerization (QAI) is the conversion
of a strained-ring reactant into a more stable product
via a photoinduced electron-transfer chain reaction
where a single photon causes isomerization of many molecules.1
The ring-opening reactions of the radical cations of
hexamethyl Dewar benzene (1) and Dewar
benzene have been studied using density functional theory
and complete active space self-consistent field (CASSCF)
calculations. The high quantum yield for ring opening
of 1 by a chain mechanism is termed
"quantum amplified isomerization" (QAI). Why QAI is
efficient for 1 but not other reactions is being studied
computationally. Two stable radical cations of 1
have been identified, along with transition states located
near avoided crossings. Conical intersections corresponding
to observed state crossings have also been located.
Ring opening in Dewar benzene and 1
occurs by formation of the radical cation followed by
a decrease in the flap dihedral. A rate-limiting Cs
transition state leads to a second stable radical cation
with an elongated transannular C-C bond and an increased
flap dihedral. A thermally-allowed conrotatory-like
pathway of Cs symmetry leads to the benzene
radical cation. The ease of oxidation of various systems
has been evaluated using adiabatic ionization energies
and electron affinities. Electron-transfer theory has
been applied to 1 to investigate the
limiting effects of back-electron transfer as related
to the unusual stability of two radical cations. Frequency-dependent
indices of refraction are computed from isotropic polarizabilities
to evaluate expected changes in optical properties between
reactants and products. Compound 1
shows greater contrast in index of refraction than the
parent system Dewar benzene. Other systems known to
undergo QAI and prospective systems for amplification
are under investigation. |
|
 |
|
| 1. Evans, T. R.; Wake, R. W.; Sifain, M. M. Tetrahedron Lett. 1973, 701-704.
|
|
| H/Vinyl Conical Intersections of
Hexatrienes as Related to the Hula-Twist Photoisomerization |
|
|
| Joseph E. Norton and
K. N. Houk
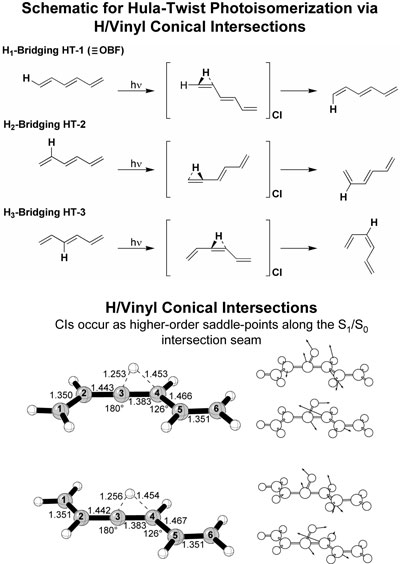
H/Vinyl Conical Intersections of Hexatrienes.
H/vinyl conical intersections are proposed to have a
potential role in the photochemical hula-twist isomerization
of hexatriene.1 The hula twist is a novel
volume-conserving isomerization mechanism for polyenes
in rigid media.2,3 Conical intersections
involving partial migration of a hydrogen and inversion
of a carbon are explored as potential pathways for hula-twist
isomerization. Complete active space self-consistent
field (CASSCF) and CASPT2 calculations with the 6-31G(d)
basis set are used to explore structures that would
potentially lead to hula-twist isomerization. These
structures are found to be geometrically ideal for a
volume-conserving process, but are energetically unfavorable.
Conical Intersections. Conical intersections
are efficient radiationless decay channels that play
a key mechanistic role in the photochemistry, chemical
kinetics, and spectroscopy of polyatomic molecules.
Conical intersections, though once thought to be rare,
have been found by Robb, Olivucci, Bernardi and their
coworkers, as well as other groups, to be essential
in the photochemistry of polyenes, azoalkanes, enones,
cyclohexadiene, cycloalkene electrocyclic ring-openings,
and retinal chromophores.4 Photochemical
reactions can involve multiple conical intersections
that lack symmetry observed in reactants and products.
Consequently, decay to the ground state can occur through
structurally and energetically different conical intersections.
|
|
 |
|
1. Wilsey, S.;
Houk, K. N. Photochem. Photobiol. 2002,76,
616-621.
2. Liu, R. S. H.; Hammond, G. S. Chem.--Eur.
J. 2001, 7, 4536-4544.
3. Liu, R. S. H. Acc. Chem. Res.
2001, 34, 555-562.
4. Robb, M. A.; Garavelli, M.; Olivucci,
M.; Bernardi, F. Rev. Comput. Chem. 2000,
15, 87-146. |
|
| Electronic Structures and Properties
of Twisted Polyacenes |
|
|
Joseph E. Norton and K.
N. Houk
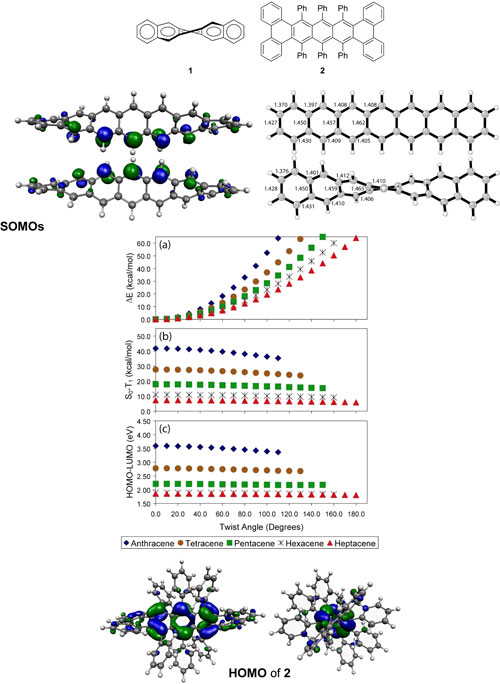
Polyacenes are linear polycyclic aromatic hydrocarbons
with properties exploitable for organic electronics. Highly
twisted polyacenes with phenyl substituents strategically
placed to induce end-to-end twisting of the acene backbone,
as illustrated by 1, have been synthesized.
Pascal et al.'s recent successful synthesis of a pentacene
exhibiting a twist of 144°, 2,1
has prompted us to investigate the effects of twisting
on the electronic structures and properties of polyacenes.
Such highly twisted molecules are expected to have unique
chiroptical properties, and have already been incorporated
into organic light-emitting diodes. The use of substituents
to overcome chemical instability while maintaining the
electronic properties necessary to serve as functional
materials in semiconducting devices is currently being
explored. In recent years, our group and others have reported
on the electronic structures of polyacenes.2,3
The electronic structures of substituted polyacenes are
currently being investigated.
The effects of twisting on the electronic structures and
properties of polyacenes have been investigated computationally
using DFT methods.4 Singlet-triplet (S0-T1)
and HOMO-LUMO gaps, and vertical S0-S1
transition energies are marginally affected as a function
of end-to-end twist angle. Heptacene remains a disjoint
radical. The large twist induced by bulky substituents
such as in 2 is predicted to have little
influence on electronic structure. |
|
 |
|
1. Lu, J.; Ho,
D. M.; Vogelaar, N. J.; Kraml, C. M.; Pascal, R. A., Jr.
J. Am. Chem. Soc. 2004, 126,
11168-11169.
2. Houk, K. N.; Lee, P. S.; Nendel, M.
J. Org. Chem. 2001, 66,
5517-5521.
3. Bendikov, M.; Duong, H. M.; Starkey,
K.; Houk, K. N.; Carter, E. A.; Wudl, F. J. Am. Chem.
Soc. 2004, 126, 7416-7417.
4. Norton, J. E.; Houk, K. N. J.
Am. Chem. Soc. 2005, 127,
4162-4163. |
|
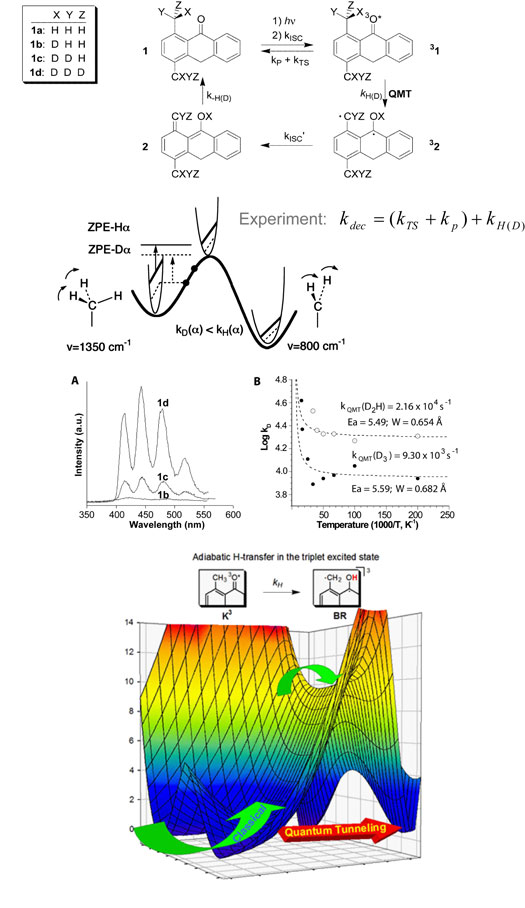
| Deuterium Tunneling in Triplet o-Methylanthrones:
Secondary Alpha Isotope Effects |
|
|
Luis M. Campos, K. N. Houk,
and M. A. Garcia-Garibay
Reactions that proceed at extremely low temperatures represent
an interesting example of chemical reactivity due to quantum
mechanical tunneling (QMT) contributions from zero-point
energy levels. o-Methylanthrones have been demonstrated
to react at cryogenic temperatures, along the triplet
manifold following photoexcitation, via QMT.
In our efforts to understand the factors that affect the
sensitivity of QMT towards variable isotopic substitution
of the ortho-methyl group, we have studied the
photochemistry of 1,4-dimethyl-10H-anthracen-9-one 1,
and its isotopologues 1b-1d.
The rates of deuterium transfer in the triplet state have
been determined by phosphorescence emission, and the tunneling
rate constants were obtained. The parent protio compound
and 1a reacted faster than our detection
timescale, but we have observed a relatively large secondary
tunneling isotope effect (TIE) of 2.3 from 1d
and 1c. Density Functional Theory calculations
were carried out in order to obtain structural information
as well as the energies of activation for the H(D)-transfer
from zero-point energy levels. |
|
 |
|
|
L. M. Campos, M. V. Warrier, K. Peterfy, K. N. Houk, M. A. Garcia-Garibay, submitted.
|
|
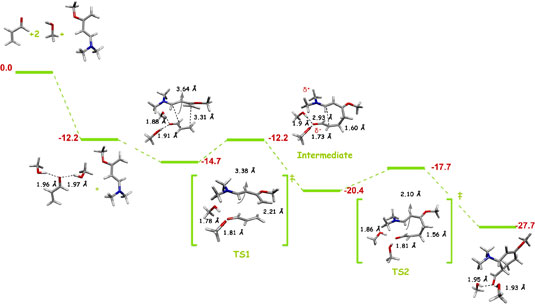
| Theoretical Study of Hydrogen Bonding
Promoted Diels-Alder Cycloadditions. Reaction Mechanism
and Origins of Enantioselectivity |
|
|
Ruth Gordillo and Kendall
N. Houk
Uncatalyzed and one MeOH molecule promoted Diels-Alder
reactions of Rawal's diene with acrolein were found to
be asynchronous but concerted processes in the gas phase.
Introduction of a second MeOH molecule produces a change
in the reaction mechanism. In this case, a stepwise mechanism
pathway, involving a zwitterionic intermediate (δ
= 0.63 e) was found. |
|
 |
|
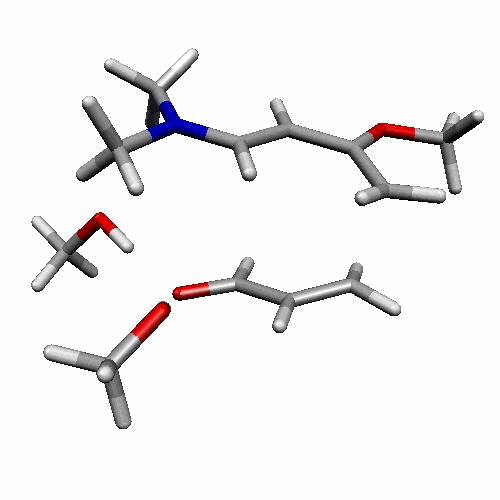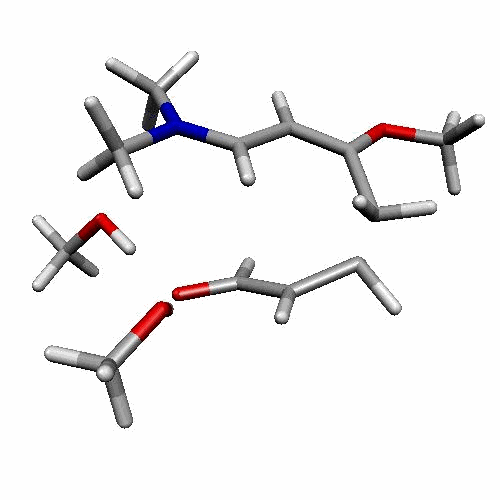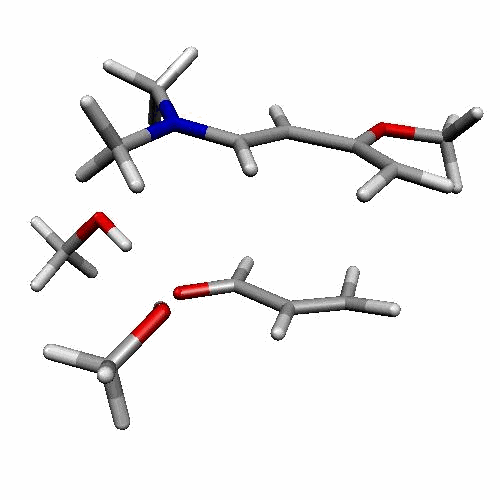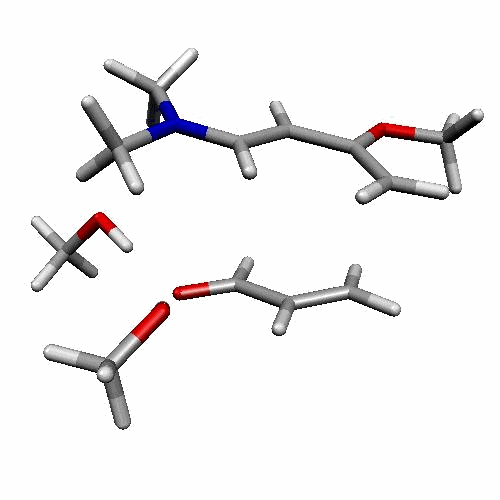
TS1 |
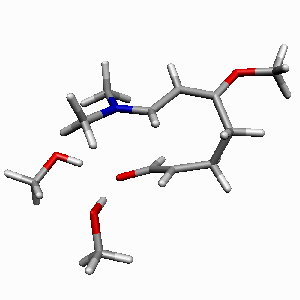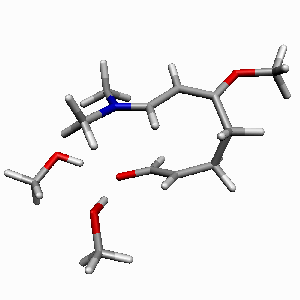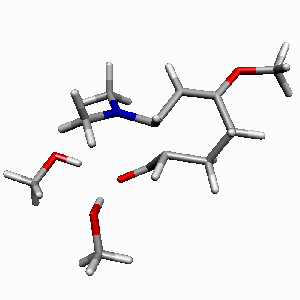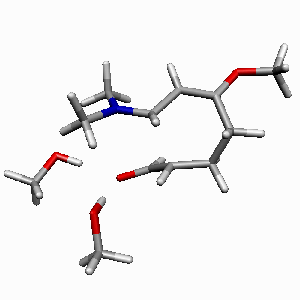
TS2 |
|
|
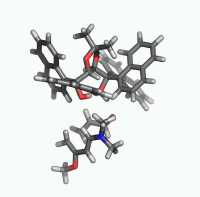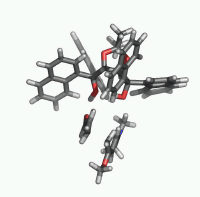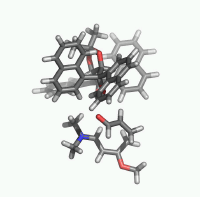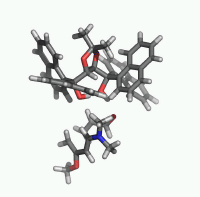
| Figure 1. Potential-energy
profile for Diels-Alder cycloaddition of Rawal's diene
with alcrolein (s-trans-endo approach
of two MeOH molecules catalyzed reaction in a bifurcated
complexation mode). ΔH energies are in kcal/mol.
Most relevant distances are shown.1,2 |
|
 |
|
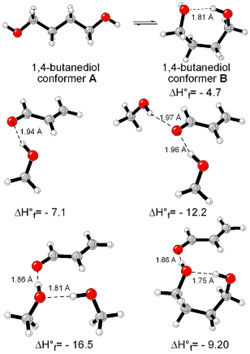
| Figure 2. 1,4-butanediol conformers
A and B optimized structures. Most stable acrolein-MeOH
complexes. In all the cases acrolein displays an s-trans
conformation. Enthalpy values are in kcal/mol. Most relevant
distances are also shown.1,2 |
|
 |
|
| Figure 3 shows one of the calculated
transition structures including the TADDOL tetranaphthyl
derivative as catalyst. The transition structure search
is being performed using a methodology that includes initial
molecular mechanic conformational search, and Morokuma's
IMOMO calculations with a combination of B3LYP-6-31G(d)
and AM1 levels of theory.3 This study will give an explanation
for the experimentally observed enantioselectivities.4
In addition, these results will provide valuable information
for improving the efficiency of structurally similar catalysts,
and for designing new organocatalysts. |
|
1. Calculations performed at the B3LYP/6-31G(d)
level of theory.
2. Ruth Gordillo, Travis Dudding, and K. N. Houk. Manuscript
in preparation.
3. Maseras, F; Morokuma, K. J. Comput. Chem. 1995, 16, 1170-1179.
4. Huang, Y.; Unni, A. K.; Thadani, A. N.; Rawal, V. H.
Nature 2003, 424, 126. |
|
| Origins of the High Affinity Binding
of the Biotin-(Strept)Avidin Complex |
|
|
Jason DeChancie and K. N.
Houk
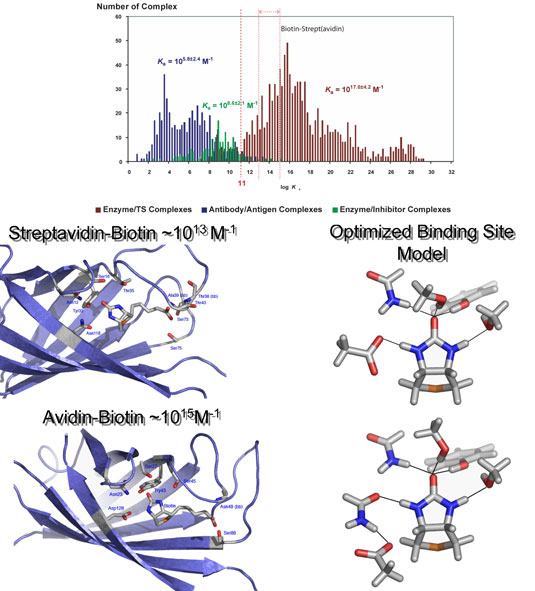
DFT and MP2 calculations employing model systems for the
(strept)avidin binding site involving the hydrogen bonding
and hydrophobic residues to the ureido moiety of biotin
have been carried out. Biotin is predicted to be significantly
polarized in the binding site. Cooperative hydrogen bonding
enhances binding compared to individual interactions.
Aspartic acid is the key residue stabilizing the hydrogen
bonding network. Aspartic acid is directly hydrogen bonded
with biotin in streptavidin and is one residue removed
in avidin, and the latter is surprisingly more favorable.
This work is an advancement towards the answer to a long
lasting question: Why are the majority of protein-ligand
interactions so poor in comparison to the biotin-(strept)avidin
complex? |
|
 |
|
| Origins of Stereoselectivity in
Allylations by Leighton's Silane Reagents |
|
|
Xiyun Zhang, K. N. Houk,
and J. L. Leighton
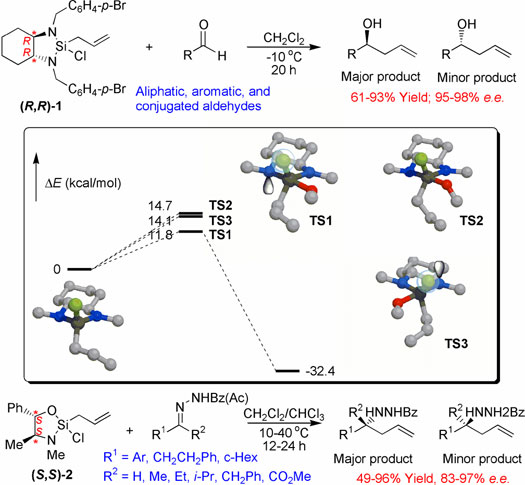
A diamine-based silane reagent (R,R)-1
has been developed for the asymmetric allylation of aldehydes.1
The mechanism and stereoselectivity of such a reaction
has been studied with MP2/6-311++G**//HF/6-31G*. The reaction
occurs in a single concerted step. Strain in the 5-membered
diazasilane ring is released upon the allylation and hence
the reaction is low-barrier and exothermic. The most favorable
transition state TS1 suggests the components
important for the stereoselectivity: 1) attack of aldehyde
O on an apical position of the Si center (anti to an N);
2) an antiperiplanar arrangement of an O lone-pair and
the Si-Cl bond in the chair transition state; 3) location
of the Cl with the lone-pair anti to the lone-pair of
apical N.2 Two less favored transition states,
TS2 and TS3, are also
shown. TS2 has an synperiplanar arrangement
of an O lone-pair and the Si-Cl bond in the chair transition
state; in TS3, Cl has its lone-pair syn to the lone-pair
of apical N.
Pseudoephedrine-based silane reagent (S,S)-2
has also been developed as a versatile reagent for the
enantioselective allylation of aldehyde-derived acetylhydrazones
and ketone-derived benzoylhydrazones.3,4 Computational
studies on such reactions are in progress. |
|
 |
|
1. Kubota, K.; Leighton,
J. L. Angew. Chem. Int. Ed. 2003,
42, 946.
2. Zhang, X.; Houk,
K. N.; Leighton, J. L. Angew. Chem. Int. Ed. 2005,
43, 938.
3. Berger, R.; Rabbat,
P. M. A.; Leighton, J. L. J. Am. Chem. Soc. 2003,
125, 9596.
4. Berger, R.;
Duff, K.; Leighton, J. L. J. Am. Chem. Soc. 2004,
126, 5686. |
|
| Mechanisms of HOONO and MeOONO Rearrangements:
A Conformation-Dependent Homolytic Dissociation |
|
|
Yi-Lei Zhao, K. N. Houk,
and Leif P. Olson
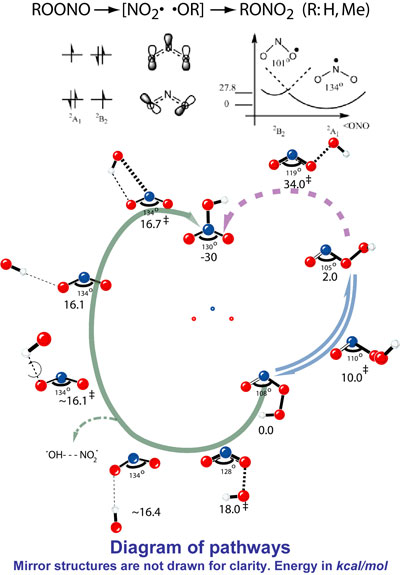
Peroxynitrite and related compounds play important roles
in biochemistry and atmospheric science. Peroxynitrite
may be formed in vivo from superoxide and nitric oxide.
The anion is fairly stable, but the protonated form decays
to nitrate in seconds at room temperature. Whether NO2
radical exists as an intermediate in the rearrangement
has been debated for decades in both experimental and
theoretical sides, since active NO2 radical
is harmful in physiology but is helpful in tropospheric
ozone formation.
Using CCSD, CCSD(T), and CBS-QB3 methods, the O-O bond
breaking process is investigated, concluding that the
OONO dihedral angle has a remarkably large influence on
barriers for the cleavage. A barrier of 18-19 kcal/mol
is predicted for RO-ONO dissociation involving a 2A1-like
NO2 fragment in TSs from a cis-conformation
(green line), while a barrier of 33-34 kcal/mol relative
to a 2B2-like TS with trans-conformation
(red line).
Notably, the favored cis-pathway is "electronically
correct" but "geometrically incorrect" for subsequent
N-O bond formation. The imperfect orientation rationalizes
some escape of free radicals, in competition with low-barrier
RO/NO2 collapse towards RONO2.
For HOONO, the pathway includes a hydrogen-bonded intermediate,
OH---ONO, earlier proposed as a source of one-electron
oxidant. MeOONO has a similar rearrangement mechanism,
but without any hydrogen-bonded intermediate. |
|
 |
|
| Thionitroxide, Formed Reversibly
by Cysteine (Protein) plus Nitric Oxide, can Serve as
a NO Reservoir or a Precursor Towards S-Nitrosothiol and
HNO |
|
|
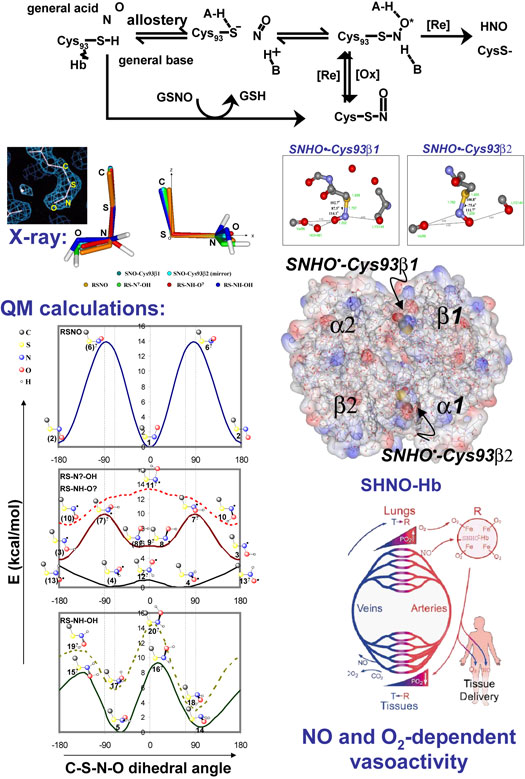
Yi-Lei Zhao and K. N. Houk
Nitric oxide (NO) plays important roles in biology. It
has been proposed that nitrosothiol (RSNOs) are key derivatives
of proteins such as hemoglobin (Hb) in erythrocyte and
formed by the reaction of specific cysteines (e.g.,
Hb-Cys93) and NO by some oxidative process. Through computational
studies with DFT, CBS, and G2 methods, we propose that
thionitroxides, RSNHO•, can be formed either from
thiols and NO or by reduction of SNO-Hb. The purported
hemoglobin-SNO in crystal structures is now thought to
be a hemoglobin-SNHO species. Formation of this polar
species benefits from hydrogen bonds; thereby conformational
processes (R/T-Hb) coupled to SNO-Hb
formation may now be explained by SNHO-Hb formation. |
|
 |
|
Crystal structures:
Chan et al. Biochemistry, 1998,
37(47), 16459-16464.
Chan et al. Biochemistry, 2004,
43(1), 118-132.
Y.-L. Zhao, K. N. Houk, submitted. |
|
| Theoretical Modeling of the Solvent
Dependence of Kinetic Resolution of Phenylethylamine with
a Chiral Amide |
|
|
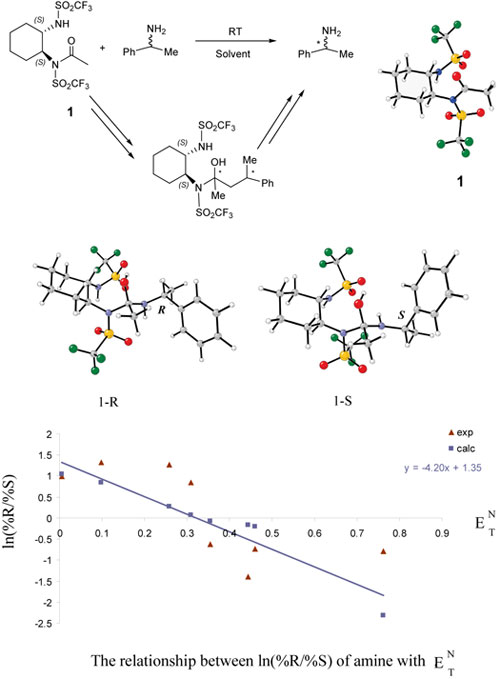
Arash Jabbari and K. N.
Houk
Substantial progress has been made in the development
of nonenzymatic acylation catalysts for the kinetic resolution
of alcohols. Less attention has been given to nonenzymatic
kinetic resolution of chiral amines. Recently Wagner and
Mioskowski reported a new and highly enantioselective
acylating reagent 1 for the resolution
of phenylethylamine and other chiral primary amines with
a unique solvent-induced reversal of stereoselectivity.
A quantum mechanical study of the origin of stereoselectivity
and the solvent polarity dependence of this reaction is
in progress. The mechanism involves a tetrahedral intermediate.
The most stable stereoisomers of this intermediate have
been computed with B3LYP/6-31G(d) in different solvents.
In the gas phase and in solvents with low polarity, the
intermediate, 1-R, with the R configuration at the indicated
carbon is the most stable form. By increasing the polarity
of the solvent, the isomer 1-S with the S configuration
becomes the most stable isomer.
The stereoselectivity correlates rather well with solvato-chromic
polarity parameters, and ENT. |
|
 |
|
| S. Arseniyadis, A. Valleix, A. Wagner
and C. Mioskowski, Angew. Chem. Int. Ed. 2004,
43, 3314. C. Reichardt, Solvents and Solvent
Effects in Organic Chemistry, 3rd ed, Wiely-VCH,
Weinheim, Germany, 2002. |
|
| Quantum Mechanical Studies of Diels-Alder
Reactions Involving Furan |
|
|
Susan N. Pieniazek and K.
N. Houk
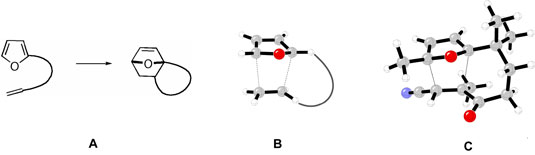
Jung and researchers utilize Diels-Alder reactions involving
furan as a diene (A, Figure 1). These reactions yield
versatile oxanorbornenes that have been used in the syntheses
of numerous complex targets.1 Substrate inertness2 and
the tendency for products to undergo retrocycloaddition3
can cause reaction failure. Exploring these reactions
with computational methods provide useful insight into
the factors controlling activation and reaction energies.
In particular, these reactions are found to be sensitive
to the nature and length of tether (B, Figure 1), as well
as substituents on the diene, dienophile, and tether (C,
Figure 1). This provides structural guidelines to design
successful reactions and to prevent cycloaddition failure.
|
|
 |
|
| Figure 1. A) Intramolecular Diels-Alder
reactions involving furan as a diene. Factors that control
the stability of transition states and products include
B) length of the tether and C) substituents on the diene,
dienophile, and tether. |
|
1. (a) Vogel, P.; Cossy, J.; Plumet,
J.; Arjona, O. Tetrahedron, 1999, 55, 13521-13642. (b)
Kappe, C. O.; Murphree, S. S.; Padwa, A. Tetrahedron,
1999, 53, 14179-14233. (c) Lipshutz, B. H. Chem. Rev.
1986, 86, 795-819.
2. Wenkert, E.; Moeller, P. D. R.;
Piettre, S. R. J. Am. Chem. Soc. 1988, 110, 7188-7194.
3. (a) Bilovic, D.; Stojanac, Z.; Hahn, V. Tetrahedron
Lett. 1964, 5, 2071-2074. (b) Kwart, H.; King, K. Chem.
Rev. 1968, 68, 415-447. |
|
|
|
|
|
|