|
|
Email: mfh@chem.ucla.edu |
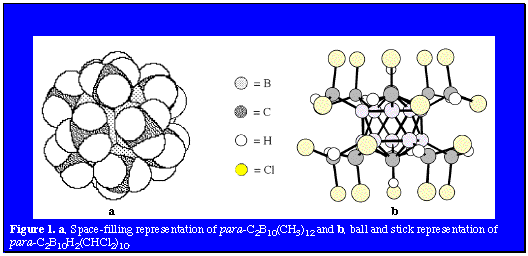
Camouflaged Polyhedral Species [pdf] The three isomers of the icosahedral C2B10H12 (1,2- or ortho-; 1,7- or meta-; and 1,12- or para-) carboranes and the isoelectronic polyhedral borane dianion B12H122- are uniquely suited to play the role of building-blocks in the construction of molecular scaffolding and stereochemically rigid platforms. These versatile components often provide sites for further functionalization of the macrostructure or to add other interesting chemical properties. This versatility has lead to the formation of several different types of structural motifs, including carborods, mercuracarborands, carboracycles, and camouflaged carboranes and polyhedral borane anions. This section of the web site will focus on the latter of these structural types, the camouflaged carboranes and polyhedral borane anions. The term camouflaged carborane or polyhedral borane describes a molecule in which all, or very nearly all, the B-H vertices have been substituted by a typical organic substituent. The resulting polyhedral borane derivative, which has a borane core stabilized by multicenter electron delocalization, is shrouded by a periphery of organic substituents. Hence, camouflaged carboranes demonstrate chemical properties indicative of their hybrid nature between that of the thermally and chemically stable carboranes and boranes and the substitutionally versatile aliphatic hydrocarbons.
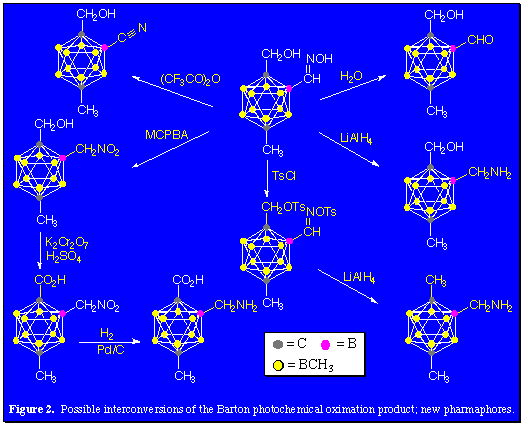
Another application which is of particular interest is the possible adoption of these camouflaged icosahedral carboranes as the bases of an inventory of large, hydrophobic and biodegradation-resistant pharmacophore groups and their potential functionalization for use in drug discovery. A comparison of the calculated van der Waals diameters of dodecamethyl-para-carborane (990 pm), decakis(dichloromethyl)-para-carborane (1440 pm) and a well-known hydrophobe, C60 (1070 pm) shows the similar volumes of these three molecules. In order to permit either of these camouflaged carboranes to be used as pharmacophore groups, reactions which provide further functionalization of the carboranes needed to be developed. The photochlorination results demonstrate that reactions characteristic of aliphatic hydrocarbons can be utilized to functionalize the organic shroud surrounding the delocalized polyhedral cage bonding of the carborane scaffolding. This hydrocarbon-like reactivity was further exploited through adaptation of the Barton photochemical oximation reaction.
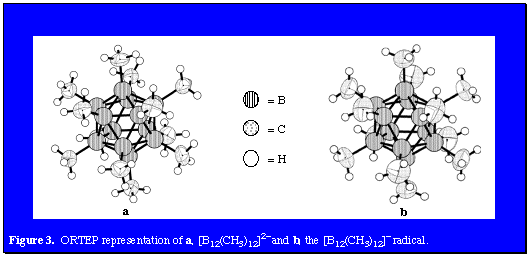
Having successfully developed a synthetic approach to decamethyl- and dodecamethyl-para-carboranes, it became feasible to attempt the similar methylation of 1,2- and 1,7-C2B10H12 and at the same time perhaps increase the yields of decamethyl- and dodecamethyl-para-carborane. A new procedure employing AlCl3 in neat CH3I solvent easily methylated para-carborane in high yield by substitution of all available B-H vertices. This procedure allowed for the preparation of 4,5,7,8,9,10,11,12-octamethyl-1,2-dicarba-closo-dodecaborane(12) and 4,5,6,8,9,10,11,12-octamethyl-1,7-dicarba-closo- dodecaborane(12) derivatives of 1,2- and 1,7-C2B10H12 after 50 h using either 1 or 10 equivalents of AlCl3. In both cases the two equivalent B-H vertices nearest the 1,2- or 1,7- pairs of C-H vertices escaped reaction. The series of neutral, monoanionic (CB11H(CH3)11- (prepared by Michl and coworkers), and dianionic camouflaged carboranes was complete with our recent report of the methylation of B12H122-. Permethylation occurred when Cs2B12H12 was first treated with a mixture of methyl iodide and trimethylaluminum (6 days, 45°C) giving Cs2B12(CH3)11I followed by the reaction of this intermediate with neat trimethylaluminum. Figure 3a presents an ORTEP representation of this extraordinary dianion. As in the cases of the permethylated carborane and monocarbon carborane anion analogs, the icosahedral surface of B12(CH3)122- presents a virtual forest of hydrophobic methyl groups even though some salts of this species display solubility in water. This amphiphilic ion may serve as a weakly coordinating dianion in a variety of applications. In addition, functionalized modifications of this species could serve as pharmacophores. Of particular interest was the fact that cyclic voltammetry in acetonitrile produced a reversible one-electron oxidation at 0.41 V. Chemical oxidation with Ce4+ ion in the same solvent led to the isolation of the bright blue, air-stable anion radical [B12(CH3)12]- in good yield (66%). Figure 3b is an ORTEP representation of this anion radical. Not surprisingly, bonding features of the diamagnetic dianion and the paramagnetic anion radical are essentially identical.
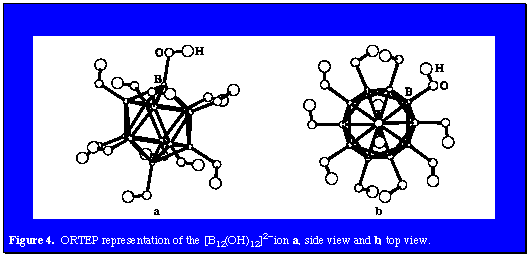
The chemistry described above exemplifies the great similarity between aromatic polyhedral borane chemistry and the aromatic branch of organic chemistry. Modular syntheses with carboranes and polyhedral borane derivatives as well as the discovery of a variety of camouflaged derivatives clearly reveals the tentacles of organic chemistry reaching into the polyhedral borane field. Thus, the conflux of boron and carbon chemistries is broadened and strengthened. Combination of this new chemistry with previously elucidated metal chemistry provides a totally manmade field of chemistry of infinite scope. Toralf Peymann, Axel Herzog, Carolyn B. Knobler, and M. Frederick Hawthorne, "Aromatic Polyhedral Hydroxyborates: Bridging Boron Oxides and Boron Hydrides," Angew. Chem. Int. Ed., 38(8), 1061, (1999). Toralf Peymann, Carolyn B. Knobler, and M. Frederick Hawthorne, "An Unpaired Electron Incarcerated Within an Icosahedral Borane Cage: Synthesis and Crystal Structure of the Blue, Air-Stable {[closo-B12(CH3)12].}- Radical," Chem. Comm., 2039 (1999). Axel Herzog, Andreas Maderna, George N. Harakas, Carolyn B. Knobler and M. Frederick Hawthorne, "A Camouflaged Nido-Carborane Anion: Facile Synthesis of Octa-B-methyl-1,2-dicarba-closo-dodecaborane(12) and Its Deboration Reaction," Chem. Eur. J., 5, 1212, (1999). Axel Herzog, Carolyn B. Knobler and M. Frederick Hawthorne, "Adaptation of the Barton Reaction to Carborane Chemistry: The Synthesis and Reactivity of 2-Hydroxyimino-1-hydroxymethylnona-B-methyl-1,12-dicarba-closo-dodecaborane(12)," Angew. Chem., 37, 1552, (1998). Wei Jiang, Carolyn B. Knobler, Mark D. Mortimer, and M. Frederick Hawthorne, "A Camouflaged Icosahedral Carborane: Dodecamethyl-1,12-dicarba-closo-dodecarborane(12) and Related Compounds," Angew. Chem., 34, 1332, (1995). View All |