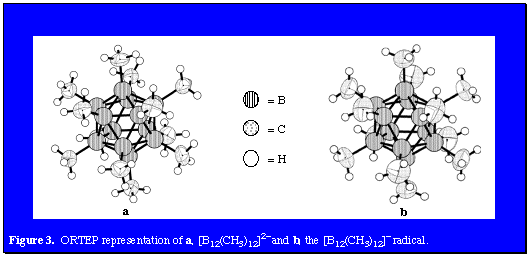
The series of neutral, monoanionic (CB11H(CH3)11- (prepared by Michl and coworkers), and dianionic camouflaged carboranes was complete with our recent report of the methylation of B12H122-. Permethylation occurred when Cs2B12H12 was first treated with a mixture of methyl iodide and trimethylaluminum (6 days, 45°C) giving Cs2B12(CH3)11I followed by the reaction of this intermediate with neat trimethylaluminum. Figure 3a presents an ORTEP representation of this extraordinary dianion. As in the cases of the permethylated carborane and monocarbon carborane anion analogs, the icosahedral surface of B12(CH3)122- presents a virtual forest of hydrophobic methyl groups even though some salts of this species display solubility in water. This amphiphilic ion may serve as a weakly coordinating dianion in a variety of applications. In addition, functionalized modifications of this species could serve as pharmacophores. Of particular interest was the fact that cyclic voltammetry in acetonitrile produced a reversible one-electron oxidation at 0.41 V. Chemical oxidation with Ce4+ ion in the same solvent led to the isolation of the bright blue, air-stable anion radical [B12(CH3)12]- in good yield (66%). Figure 3b is an ORTEP representation of this anion radical. Not surprisingly, bonding features of the diamagnetic dianion and the paramagnetic anion radical are essentially identical.
The methyl-camouflaged species described above demonstrate the amplification of aromatic polyhedral borane chemistry by the prudent application of organic chemistry and the emergence of a novel family of organoboranes, the camouflaged carboranes and polyhedral boranes. The development of successful routes for the electrophilic polymethylation of icosahedral carboranes and the [B12H12]2- ion provides species equipped with hydrophobic hydrocarbon surfaces, thereby spawning the field of hydrophobically-camouflaged carboranes and polyhedral boranes. As a consequence, the possible existence of similar structures presenting hydrophobically-camouflaged surfaces offered a synthetic challenge. While the oxidation of boranes is highly exothermic and reminiscent of high-performance aircraft fuels, rocket propellants, and explosives, moderation of this reaction using relatively weak oxidants with stabilized icosahedral borane derivatives offered a challenging pathway to polyhydroxylated products. Our approach proved to be successful using 30% hydrogen peroxide as the solvent and oxidant at the reflux temperatures with B12H122-, CB11H12- and 1,12-(CH2OH)2-1,12-C2B10H10 as substrates. Figure 4 presents the structure of B12(OH)122-. In each case all B-H vertices were converted to their B-OH counterparts in variable yield, Cs2[closo-B12(OH)12] 65%, Cs[closo-1-H-1-CB11(OH)10] 31%, and closo-1,12-H2-1,12-C2B10(OH)10 80%, respectively.
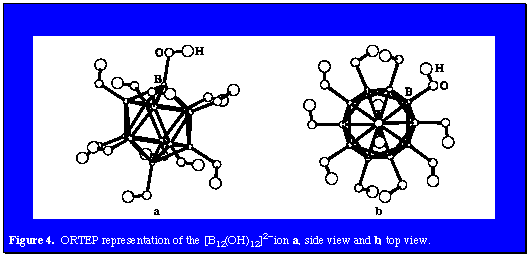
The properties of the [B12(OH)12]2- ion are of particular interest since this structure offers a unique geometric platform for formation of extended structures such as dendrimers. An unexpected property is the facile aggregation of the disodium salt which has very limited water solubility. Sodium ions coordinate with the -OH surface and provide intercage bonding. The dicesium salt is freely soluble in water since cesium is too large to effectively coordinate with the [B12(OH)12]2- ion. While [B12(OH)12]2- may be viewed as an intermediate in the hydrolysis of [B12H12]2- to boric acid, it still retains aromatic character and twenty-six cage-bonding electrons available for chemical reduction reactions.
Page 1 2 3 4 
|